KOVA Korean Variant Archive for a reference database of genetic variations in the Korean population
Cohorts and sample preparation
We collected WES and WGS data of Korean individuals from independent research groups in Korea. All sequencing data were obtained from normal tissues or blood samples following standard protocols. This project was performed with approval of the Institutional Review Board of each group (Seoul National University and others), in which all donors provided a written informed consent. All the experiments were performed in de-identified status and in accordance with relevant guidelines and regulations.Variant calling and filtering
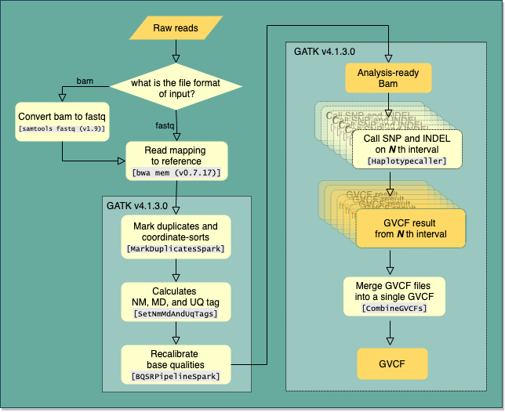
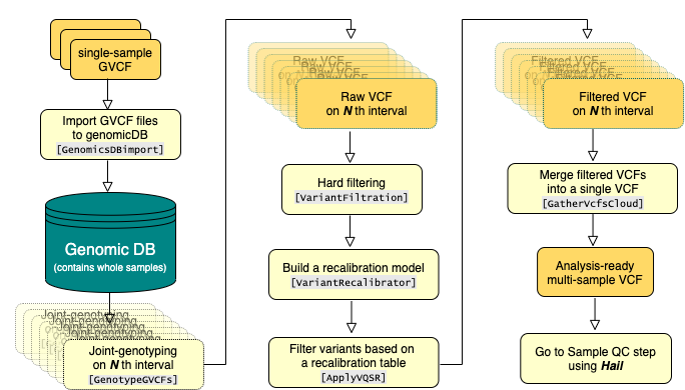
We used BWA mem with default options to align raw read sequences to the GRCh38+decoy reference sequence. After marking duplicates and sorting by coordination with MarkDuplicatesSpark, the mapping quality was recalibrated by BQSRPipelineSpark which is implemented on GATK version 4.1.3.0. Single nucleotide variants and small indels were then called using GATK HaplotypeCaller with option ‘-ERC GVCF’ throughout the intervals we defined. To genotype samples jointly, we created genomic DB and followed GATK best practice guideline. SNVs and Indels were recalibrated by GATK’s VQSR model to select 99.7% of true sites from the training set. Because we called WES and WGS variants separately, we merged the variants using hail. We then excluded multi-allelic variants and variants with GQ <20 or DP<10 or AB<0.2. Variants which violate Hardy-Weinberg equilibrium on allelic frequency (P < 10-6) when allele frequency is above 0.01 or inbreeding coefficient score is less than -0.03 were also removed. Functional annotation was performed by Variants Effect Predictor (VEP) version 101. Most severe consequences per gene were selected for each variant using gnomAD python library.