
KoNA data format
- KoNA data format : [download]
KoNA was built based on INSDC's data format and structural model. It provides genome data storage services to researchers in Korea and worldwide, similar to major countries. Researchers who have submitted data in INSDC databases such as SRA in the U.S or ENA in Europe or have experience using a search service will quickly understand the data structure of KoNA
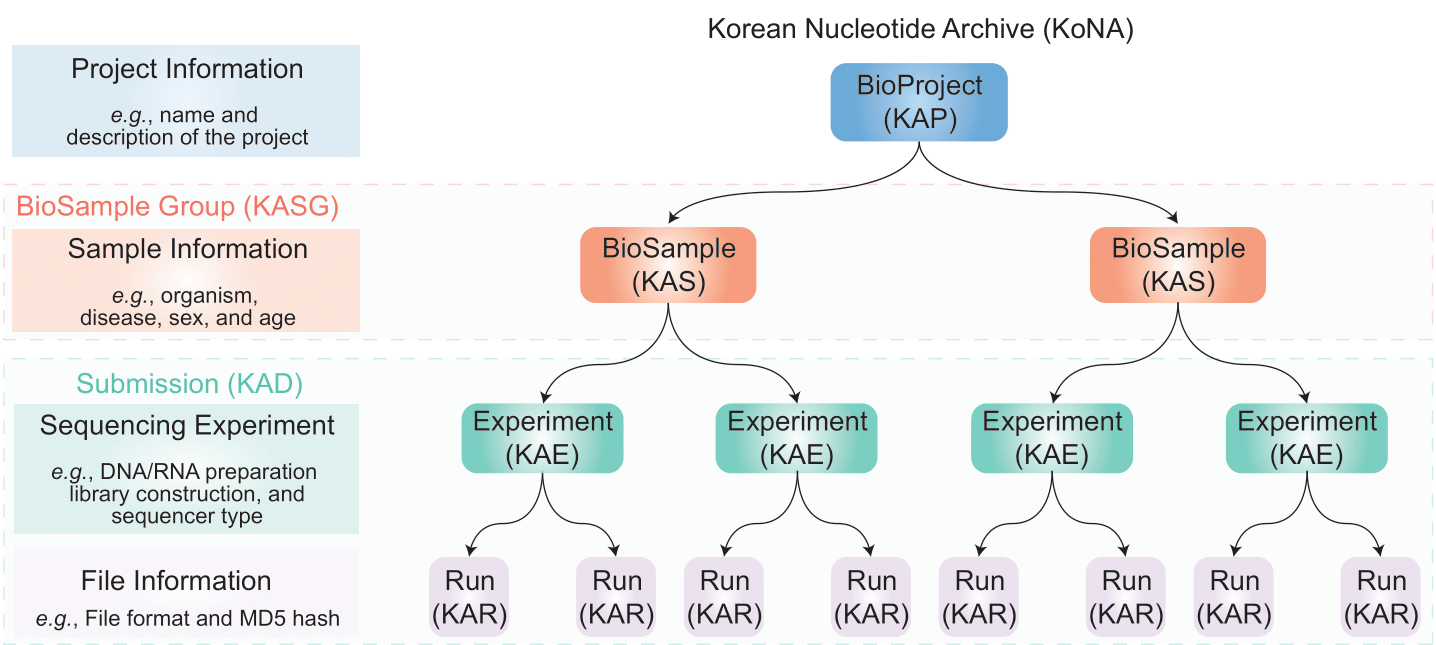
Genome data stored in KoNA is divided mainly into raw data and metadata. Raw data are genome reads produced through sequencing devices, including FASTQ files. Metadata is information that describes raw data and primarily consists of Project, Sample, Experiment, and Run. This format is the same as that of the INSDC database.
Metadata format
Metadata primarily consists of Project, Sample, Experiment, and Run, which are related. In other words, to submit raw data, project-sample-experiment-run information must be present and linked with each other.
Project information is implemented as a BioProject database, including descriptions of the project and study information for genome production. BioProject contains information such as the submitter, date, project design, and publication. BioProject is independent and is not linked to other data but can be linked to an umbrella project. In addition, if there are any BioProjects that may be included in large-scale projects or consortiums (e.g., Korea Post-Genome Project), the BioProjects contain information on their umbrella projects. The BioProject accession IDs start with “KAP.”
Sample information is implemented as a BioSample database and includes information on biological source materials or samples used for data production. BioSample must be structurally linked with BioProject. Therefore, prior to submitting sample information in BioSample, project information must be submitted in BioProject and approved by KoNA administrator (i.e., submission of BioSamples requires their ‘parent’ BioProject accession ID). To submit several BioSamples of the same type (e.g., samples of several lung cancer patients), BioSample Group will be necessary. The submitters may use a spreadsheet to describe information about their BioSamples: each row in the spreadsheet stands for each BioSample, and the spreadsheet itself stands for the BioSample Group. The accession ID of BioSample Group starts with “KASG,” and the accession ID of each BioSample starts with “KAS.”
Each experiment represents a unique sequencing library for a specific sample, and experiment and run are grouped with Korea Read Archive (KAD).
Four types of metadata objects
 <Relationships between BioProject, BioSample Group, and KAD>
BioProject, BioSample Group, and KAD data are linked as shown above. Metadata objects of BioProject do not contain linkage information with other data except umbrella projects. Therefore, BioProject is independent. BioSample data must have linkage information with BioProject, and allow one to gain project information from which the sample originated. KAD must have information about the linked BioSample Group. As such, BioProject, BioSample Group, and KAD are linked with each other.
Raw data format
Raw data is the data produced by sequencing devices (i.e., Illumina sequencer). We recommend the submitters to remove contaminant reads (i.e., adapter sequences) before the submission of the raw data. The representative raw data format is the FASTQ file format (exceptionally, for the data generated by some long-read sequencing platforms, such as PacBio, we also allow FASTA format):
FASTQ format is a representative format for genome read. This text-based form is to score biological reads expressed as “AGCT (uppercase)” or “acgt (lowercase)” of the nucleotide sequence and quality (Phred) score for each nucleotide indicated by ASCII code letters.
<Relationships between BioProject, BioSample Group, and KAD>
BioProject, BioSample Group, and KAD data are linked as shown above. Metadata objects of BioProject do not contain linkage information with other data except umbrella projects. Therefore, BioProject is independent. BioSample data must have linkage information with BioProject, and allow one to gain project information from which the sample originated. KAD must have information about the linked BioSample Group. As such, BioProject, BioSample Group, and KAD are linked with each other.
Raw data format
Raw data is the data produced by sequencing devices (i.e., Illumina sequencer). We recommend the submitters to remove contaminant reads (i.e., adapter sequences) before the submission of the raw data. The representative raw data format is the FASTQ file format (exceptionally, for the data generated by some long-read sequencing platforms, such as PacBio, we also allow FASTA format):
FASTQ format is a representative format for genome read. This text-based form is to score biological reads expressed as “AGCT (uppercase)” or “acgt (lowercase)” of the nucleotide sequence and quality (Phred) score for each nucleotide indicated by ASCII code letters.
<Relationships between BioProject, BioSample Group, and KAD>
BioProject, BioSample Group, and KAD data are linked as shown above. Metadata objects of BioProject do not contain linkage information with other data except umbrella projects. Therefore, BioProject is independent. BioSample data must have linkage information with BioProject, and allow one to gain project information from which the sample originated. KAD must have information about the linked BioSample Group. As such, BioProject, BioSample Group, and KAD are linked with each other.
Raw data format
Raw data is the data produced by sequencing devices (i.e., Illumina sequencer). We recommend the submitters to remove contaminant reads (i.e., adapter sequences) before the submission of the raw data. The representative raw data format is the FASTQ file format (exceptionally, for the data generated by some long-read sequencing platforms, such as PacBio, we also allow FASTA format):
FASTQ format is a representative format for genome read. This text-based form is to score biological reads expressed as “AGCT (uppercase)” or “acgt (lowercase)” of the nucleotide sequence and quality (Phred) score for each nucleotide indicated by ASCII code letters.
@<identifier and expected information>
<sequence>
+<identifier and other information OR empty string>
<quality>
- Line 1 is the same as the title of the sequence that starts with “@” and contains a description of the sequence.
- Line 2 is the nucleotide text.
- Line 3 starts with “+,” followed by the same description as in line 1.
- Line 4 is the ASCII code, encoding the quality score for each nucleotide. The same number of codes as the nucleotide text must be inputted.
- 1. When forward and reverse sequences are submitted as separated files, the sequences in each file must appear in the same order.
- 2. If both forward and reverse reads are submitted in the same file, the reads must alternate and written in eight lines. For example, “1R” must come after “1F,” followed by “2F” and “2R.”
@HWUSI-EAS100R:6:73:941:1973#0/1
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+HWUSI-EAS100R:6:73:941:1973#0/1
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65