- 카테고리 Transcriptomics > RNA-seq-transcriptomics
- 수정일2025-10-31 18:37:00
- 레퍼런스
RNA-seq Analysis Pipeline은 RNA-Seq 데이터를 처리하고 유전자 발현에 대한 통계적 분석을 수행하는 것을 목표로 합니다. 이 파이프라인은 유전자의 발현 수준을 이해하고 해석하기 위해 실험 데이터의 품질을 평가하고 정제하며, 정렬, 발현 수준 계산, 통계적 분석, 결과 시각화로 구성되어 있습니다.파이프라인의 초기 단계에서는 실험 데이터의 품질 평가와 정제가 이루어집니다. FastQC를 사용하여 실험 데이터의 품질을 검사하고 평가하며, Cutadapt를 활용하여 시퀀싱 어댑터 및 낮은 품질의 리드를 효과적으로 정제합니다.다음으로, STAR 2를 이용하여 리드를 정확하게 유전체에 정렬하고, 각 유전자의 발현 위치를 정밀하게 파악합니다. 그 후, Rsubread 라이브러리의 FeatureCounts를 활용하여 정렬된 리드를 각 유전자에 할당하여 발현 수준을 정량화하고 Count Matrix를 생성합니다.이어지는 단계에서는 R 기반의 edgeR과 limma를 사용하여 발현 수준의 통계적 차이를 식별하고 각 유전자의 발현 변동을 분석합니다. 이 과정에서 control과 test 샘플에는 각각 최소 두 개 이상의 생물학적 복제 샘플이 포함되어야 통계적 분석이 가능하다는 점에 유의해야 합니다. 복제가 없는 경우 잔차 유도가 0이 되어 분석이 실패하거나 결과가 신뢰성을 잃을 수 있습니다. 또한, R 기반의 fgsea를 활용하여 gene set 간의 풍부도를 평가하고, 다양한 시각화 도구를 활용하여 효과적으로 표현합니다. 마지막으로, fgsea의 결과 파일을 이용하여 여러 R 패키지를 통해 데이터 시각화, 그래픽 생성 등 실험 결과를 자세히 분석하고 시각화합니다.전체적으로, 최상위 입력 데이터인 fastq 형식의 RNA-seq raw data로부터 시작하여 품질 보고서인 fastqc.report.html을 생성하고, MA plot, correlation plot, network, volcano plot, heatmap 등의 다양한 시각화 자료를 통해 유전자 발현 및 풍부도를 시각적으로 확인할 수 있습니다.
Bio-Express RNA-seq Alternative-splicing Pipeline (이하 AS)은 유전자 발현의 전사체 수준에서 크게 5가지 type의 splicing 양상을 확인할 수 있다.Alternative splicing은 하나의 유전자를 구성하는 복수의 exon (coding region)간의 조합에 따라 여러 transcripts (isoforms)가 생성되며, 이에 따라 하나의 유전자라도 서로 다른 구조를 갖는 단백질이 만들어짐에 따라 기능이 다른 유전자로써 역할을 하게된다. 이러한 메커니즘을 통해 단백질의 폭넓은 capacity 를 확보할 수 있으며, 다양한 분자적 역할이 가능하다.

(출처: From Wikipedia, the free encyclopedia)
기본적인 5가지 형태의 이벤트는 아래와 같다.
1. SE (Exon skipping | cassette exon)
2. MXE (Mutually exclusive exons)
3. A5SS (Alternative donor site)
4. A3SS (Alternative acceptor site)
5. RI (Intron retention)
.png)
AS 분석 파이프라인은 아래와 같은 흐름으로 진행됨.
1. Quality Control, 시퀀싱 품질 관리 (by FastQC)
2. Trimming, 아답터 및 low quality 제거 (by Cutadapt, Trimmomatic)
3. Mapping, 레퍼런스 alignment (by STAR, HISAT2)
4. AS detection, 선택적 스플라이싱 탐색 (by rMATs)
5. Visualization, AS 결과 시각화 (by ggsashimi)
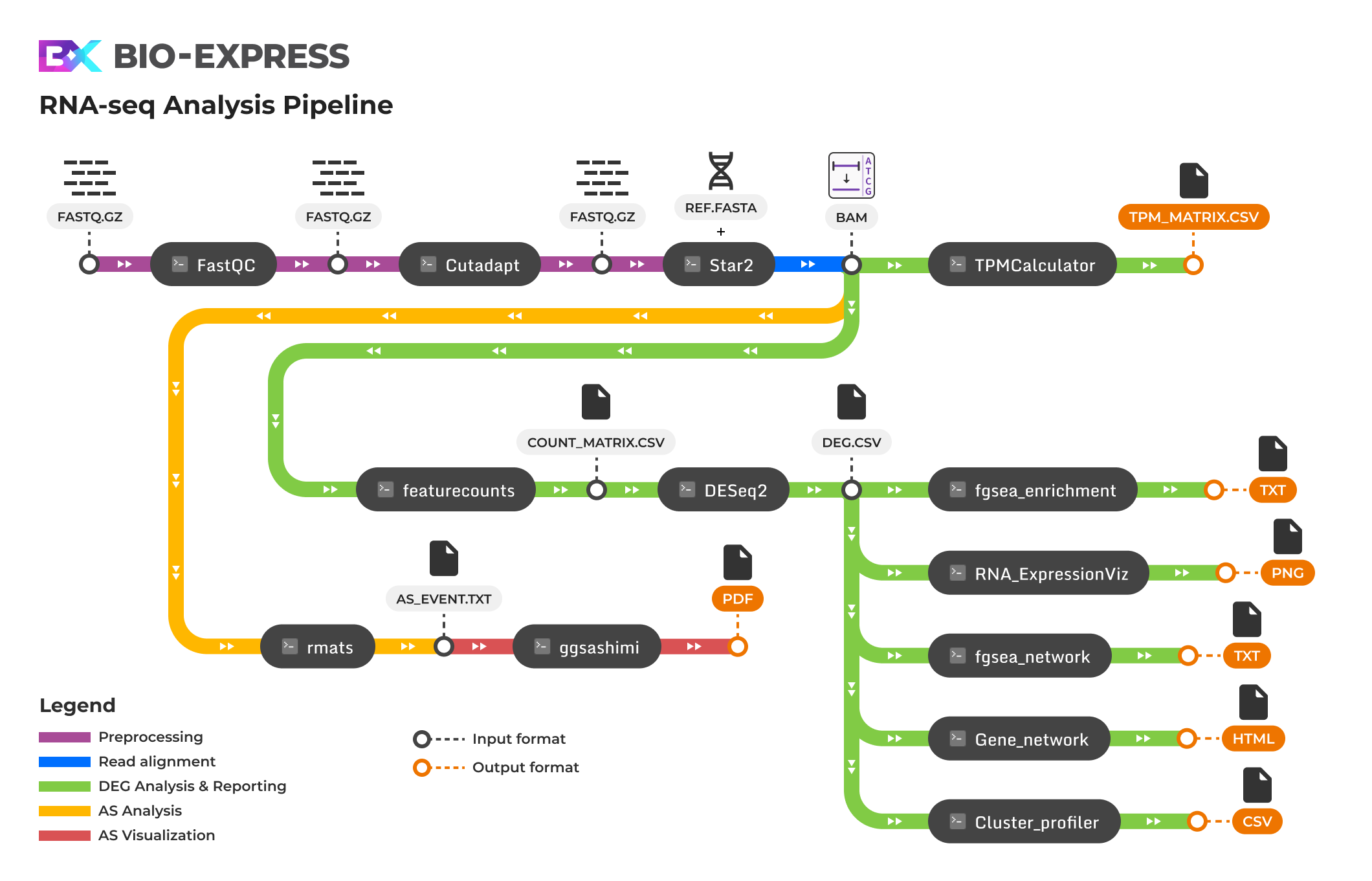
파이프라인 모듈
FastQC
FastQC는 고속 염기서열 분석(high throughput sequence) 데이터의 품질 관리를 위한 분석도구입니다. 이 프로그램은 FASTQ 형식의 서열 데이터를 읽어들여 여러 품질 관리(Qaulity Control) 검사를 수행하고 결과는 HTML 기반의 보고서로 출력합니다. FastQC는 전반적인 품질 문제에 대한 개요 정보를 제공하며, 쉽게 확인할 수 있는 요약된 그래프와 테이블을 포함합니다. FastQC는 FASTQ 형식의 파일이 입력 파일로 사용되며, 출력 결과는 리포트 html 파일과 zip 형식의 압축 파일이 생성됩니다.
주요사항
- FastQC는 자바 애플리케이션입니다. 실행하기 위해서는 시스템에 적절한 자바 실행 환경(Java Runtime Environment, JRE)이 설치되어 있어야 합니다. 따라서 FastQC를 실행하기 전에 먼저 적절한 JRE가 설치되어 있는지 확인해야 합니다. 다양한 종류의 JRE를 사용할 수 있지만, 저희가 테스트해본 것은 최신 오라클 런타임 환경과 adoptOpenJDK 프로젝트의 JRE입니다. 64비트 JRE를 다운로드하여 설치하고, 자바 애플리케이션이 시스템 경로(path)에 포함되도록 설정해야 합니다(대부분의 설치 프로그램이 이를 자동으로 처리해줍니다).
실행 명령어 예시
$program_dir/fastqc –t 6 –o $OUTPUT_DIR $INPUT_DIR/$READ
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o) |
결과
-
.png)
Basic Statistics 테이블은 주어진 FASTQ 파일에 대한 간단한 통계적 정보를 제공합니다. 일반적으로 다음과 같은 정보를 포함합니다. Filename : 분석된 파일의 이름 또는 경로 File type : FASTQ 파일의 종류 Encoding : 품질 점수 인코딩 방식 Total Sequence : 총 서열 수 Filtered Sequences : Read 품질이 좋지 않은 서열 수 Sequence length : 서열의 길이 %GC : 서열에서의 GC 백분율
-
.png)
X축은 리드의 염기 위치를 나타내며, Y축은 품질 점수를 의미합니다. 점수가 높을수록 품질이 좋습니다. 중앙의 빨간색 선은 중앙값을 나타내고, 노란색 박스는 사분위간 범위(25~75%)를 의미합니다. 위쪽 및 아래쪽의 위스커(whisker)는 각각 10% 및 90% 포인트를 나타냅니다. 파란색 선은 평균 품질을 의미합니다. 어떤 염기의 하위 사분위수가 10 미만이거나 중위값이 25보다 작을 경우 경고로 간주됩니다. 또한, 어떤 염기의 하위 사분위수가 5 미만이거나 중위값이 20보다 작으면 오류로 간주됩니다.
-
.png)
색을 이용하여 각 타일의 품질을 나타내며, 파란색은 품질이 높음을 나타내고 빨간색은 품질이 낮음을 나타냅니다. 각 타일의 품질을 모든 타일의 평균 품질과 비교하여 예상 패턴과의 편차를 식별 할 수 있습니다. 특정 타일의 품질이 지속적으로 좋지 않으면 물리적 결함이나 오염 등 셀의 특정 영역에 문제가 있음을 나타낼 수 있습니다. 이상적으로는 모든 타일이 높은 품질을 보여야 하면 플롯에서 더 차가운 색상으로 표시됩니다. 이 플롯은 Illumina 라이브러리에서만 나타납니다.
-
.png)
X축은 리드의 전체 길이에 대한 평균 품질 점수를 나타내고, Y축은 해당 품질 점수를 갖는 읽기의 수를 나타냅니다. 시퀀스의 시퀀싱 품질은 해당 시퀀스에 대한 정확성과 신뢰성을 나타내며, 품질 점수가 높을수록 오류 발생 가능성이 낮다는 것을 의미합니다. 만약 시퀀싱 실행이 전반적으로 낮은 품질을 보인다며, 시퀀싱 화학 문제나 샘플 준비 문제 등이 있을 수 있습니다. 품질 점수가 기록되지 않은 BAM/SAM 파일의 경우 확인할 수 없습니다. 품질 점수가 Phred 척도 기준 최고 품질 점수 27점(오류율 0.2%) 미만일 경우 경고 발생, 20점(오류율 1%) 미만일 경우 오류입니다.
-
.png)
X축은 리드의 포지션을 나타내고, Y축은 시퀀싱한 리드에서 각 base의 전체 비율을 나타냅니다. 좋은 품질의 시퀀싱 샘플에서는 각 위치의 염기 비율을 나타내는 4개의 선이 평행하고 서로 가까워야 합니다. 그러나 선이 일부 위치에서 엉키거나 얽히면 과도하게 표현된 시퀀스가 오염되었음을 나타낼 수 있습니다. 또한, A/T 또는 G/C 염기의 비율이 어떤 위치에서는 10% 이상 차이나면 경고 발생, 20%를 초과하면 오류입니다.
-
.png)
시퀀스에서 G와 C 뉴클레오티드의 백분율 비율을 나타냅니다. 이를 통해 DNA 또는 RNA 시퀀스의 특성을 이해할 수 있습니다. 시퀀스의 GC 함량은 DNA 안정성, 서열의 물리적 특성, 유전자 발현에 영향을 미칠 수 있으므로 중요한 지표 중 하나입니다. X축은 GC contents의 비율을 나타내고, Y축은 시퀀스의 총량을 나타냅니다. 정규 분포와 편차 합계가 전체 리드의 15%를 초과하면 경고, 30%를 초과하면 오류입니다.
-
.png)
시퀀싱 리드의 각 위치에서 발견된 N 비율을 나타내며 일반적으로 매우 낮습니다. 그러나 어떤 위치에서는 N 비율이 5%를 초과하면 시퀀싱 시스템에 문제가 있을 수 있다는 경고로 간주되며, 20%를 초과하면 오류로 간주됩니다. 데이터의 품질이 높은지 확인하고 시퀀싱 읽기의 정확성에 영향을 줄 수 있는 문제를 식별하려면 시퀀싱 중에 N 비율을 모니터링하는 것이 중요합니다. N 비율이 권장 임계값을 초과하는 경우 문제의 심층적인 분석이 필요할 수 있습니다.
-
.png)
시퀀싱 데이터에서 각 리드의 길이에 대한 분포를 나타냅니다. 이 그래프의 X 축은 시퀀스 길이를, Y 축은 리드 수를 나타냅니다. 시퀀싱 데이터에서 발견된 리드의 길이가 어떻게 분포되어 있는지를 시각적으로 확인할 수 있습니다. 이 그래프를 통해 시퀀싱 데이터 세트의 리드 길이 분포를 파악할 수 있으며, 예상치 못한 리드 길이나 이상한 분포를 감지하여 데이터의 품질을 평가하는 데 도움이 됩니다. 일반적으로 시퀀스 길이 분포는 일정하거나 특정한 패턴을 따르지만, 비정상적인 분포는 시퀀싱 데이터에 문제가 있을 수 있다는 신호일 수 있습니다.
-
.png)
중복된 시퀀스가 전체의 20% 이상일 경우 경고, 50% 이상일 경우 오류입니다.
-
.png)
시퀀싱 데이터에서 빈번하게 등장하는 시퀀스를 나타내는 테이블입니다. 이 테이블은 시퀀싱 데이터에서 특정 시퀀스가 기대보다 더 자주 나타나는 경우를 식별합니다. 실험과정에서 발생한 오류, PCR 이중성, 어댑터 오류 또는 샘플의 비정상적인 특정으로 인해 발생할 수 있습니다. 해당 내용을 확인하고 잠재적인 문제를 식별하는 것은 시퀀싱 데이터의 정확성과 신뢰성을 높이는 데 도움이 됩니다.
-
.png)
시퀀싱 데이터에서 발견된 어댑터 시퀀스의 누적 백분율을 보여주는 차트입니다. 시퀀스가 발견된 위치에 따라 백분율이 증가하며, 시퀀스가 리드의 끝까지 존재하는 동안 계산됩니다. 어댑터의 비율을 확인하여 데이터의 품질을 평가하며, 어댑터 시퀀스가 발견되는 비율이 높을수록 시퀀싱 데이터에 오류가 포함될 가능성이 높아지므로, 5%를 초과하면 경고, 10%를 초과하면 오류입니다.
Cutadapt
Cutadapt는 NGS 시퀀싱 데이터에서 어댑터(adapter) 서열 제거, 품질이 낮은 서열 제거 등 다양한 전처리 작업을 수행하는 데 사용되는 도구입니다. 주로 서열의 3’ 말단에서 염기서열을 제거하며, 특히 RNA 시퀀싱 또는 DNA 시퀀싱 데이터에서 어댑터 제거에 빈번하게 활용됩니다. 전체 데이터의 품질을 유지하여 이후의 분석 단계에서 정확한 결과를 얻을 수 있도록 도움을 줍니다. 입력 데이터로는 fastq, fastq.gz, fq, fq.gz 확장자를 가진 파일을 사용할 수 있습니다. 출력으로는 파일 확장자 앞쪽에 ‘_trimmed’라는 문구가 붙은 fastq 파일이 생성되도록 세팅되어 있습니다. 따라서 "trimmed.fastq" 파일은 프라이머 잔여물이나 다른 불순물이 제거된 reads 파일을 나타냅니다.
주요사항
- 1. Bio-Express의 Cutadapt 모듈은 paired-end와 single-end 형식의 FASTQ 데이터를 모두 처리할 수 있도록 설계되었습니다.
- 2. -a 및 –A 옵션에 입력해야 할 각각의 어댑터 서열은 NGS 시퀀서 플랫폼별로 다음과 같습니다. > Illumina: 기본값 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT > MGI: 필요시 기본값 대체 입력 -a AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA -A AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG
실행 명령어 예시
$PROGRAM_DIR/cutadapt -q 20 –Q 20 -j 6 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 --pair-filter=$PAIR_FILTER -o $OUTPUT_DIR/$READ_1 –p $OUTPUT_DIR/$READ_2 $INPUT_DIR/$READ_1 $INPUT_DIR/$READ_2
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o, -p) | |
| Option | Integer | min_len_r1 | 70 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read1의 최소 길이 | |
| Option | Integer | min_len_r2 | 1 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read2의 최소 길이 | |
| Option | String | pair_filter | any | --pair-filter (single-end FASTQ에는 미적용) - any: 두 Read 중 어느 하나라도 조건에 부합하면 필터링 - both: 두 Read 모두 조건에 부합하면 필터링 | |
| Option | String | adapter_pos | adapter | 어댑터 시퀀스 처리 위치 지정 --adapter: 서열의 3' 끝 방향에서 어댑터를 찾아 해당 어댑터와 그 이후 모든 서열을 제거 (-a, -A) --front: 서열의 5' 시작 방향에서 어댑터를 찾아 해당 어댑터와 그 이전 모든 서열을 제거 (-g, -G) --anywhere: 5' 또는 3' 어느 쪽에든 나타날 수 있는 어댑터를 탐지 (-b, -B) | |
| Option | String | adapter_r1 | AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | 첫 번째 Read의 어댑터 시퀀스 (-a) | |
| Option | String | adapter_r2 | AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT | 두 번째 Read의 어댑터 시퀀스 (-A) | |
| Option | Integer | quality_r1 | 20 | 첫 번째 Read의 절단 기준으로 사용할 품질 임계값 (-q) | |
| Option | Integer | quality_r2 | 20 | 두 번째 Read의 절단 기준으로 사용할 품질 임계값 (-Q) |
결과
-
.png)
총 입력된 read pair 수, 어댑터가 검출된 read의 수와 비율, 필터링 후 최종적으로 출력된 read 수 등 전체 리드 처리 현황을 요약함. 또한 데이터 전처리 단계에서 몇 %의 리드가 어댑터에 의해 잘렸고, 몇 %가 최종 분석에 사용 가능한지, 데이터 손실이 얼마나 발생했는지 파악함.
-
.png)
어댑터 서열 (Illumina adapter 등), 탐지 및 제거된 횟수, 최소 overlap 길이, mismatch 허용 개수 (error tolerance), 제거된 어댑터 서열에서의 염기 비율(어댑터 클리핑의 정확성 평가에 활용) 등 Read1에서 탐지된 어댑터의 정보. Read2에 대한 내용도 동일한 방식으로 출력함.
STAR2_index
STAR를 사용하기 전에 참조 게놈에 대한 인덱스를 미리 만들어야 합니다.
이 인덱스는 정렬 과정에서 참조 게놈을 효율적으로 검색하기 위한 데이터 구조입니다.
인덱스 생성 프로세스는 일반적으로 한 번만 수행되며, 생성된 인덱스는 동일한 참조 게놈을 사용하는 모든 정렬 작업에 재사용할 수 있습니다.
주요사항
- star2_index는 STAR 정렬기에 쓰는 유전체 인덱스(Genome/SA/SAindex+SJ 테이블 묶음)로, RNA-seq 리드를 빠르게 매핑·스플라이스 검출할 때 필요하며 만들 때 genomeGenerate로 FASTA+GTF를 넣고 sjdbOverhang=읽기길이-1(예: 100bp→99)로 생성; 사람 기준 RAM 수십 GB와 큰 디스크가 필요하다. 또한 참조·주석 버전 변경, 리드 길이 변화, STAR 버전 업그레이드 시엔 인덱스를 재생성하는 게 안전하다.
실행 명령어 예시
$program_dir/star2_index $OUTPUT_DIR $INPUT_DIR/$READ
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | fasta_dir | path/to/fasta_dir | STAR를 사용할 때는 하나 이상의 FASTA 파일로 구성된 게놈 참조 서열을 지정할 수 있으며, 각 파일에는 여러 개의 참조 서열(염색체라고 함)이 포함될 수 있습니다. 염색체 이름은 chrName.txt 파일을 통해 변경할 수 있으며, 파일 내의 염색체 순서는 유지되어야 합니다. 이 파일의 이름은 모든 출력 정렬 파일(.sam 등)에 사용됩니다. 염색체 이름에는 탭이 허용되지 않으며 공백 사용은 권장되지 않습니다. FASTA 파일이 여러 개 필요한 경우 FA 형식 파일이 포함된 디렉토리 경로가 입력으로 사용되며, 선택한 디렉토리 내의 모든 FA 유형 파일이 사용됩니다. 단일 파일만 선택해야 하는 경우 해당 디렉토리에 사용할 FA 유형 파일은 하나만 배치해야 합니다. | |
| Input | File | gtf_file | path/to/gtf_file | 주석이 달린 스크립트가 있는 파일의 경로를 표준 GTF 형식으로 지정합니다. STAR는 이 파일에서 스플라이스 접합을 추출하고 이를 사용하여 매핑의 정확도를 크게 향상시킬 것입니다. 이것은 선택 사항이며 STAR는 주석 없이 실행할 수 있지만, 주석이 있을 때마다 주석을 사용하는 것이 매우 권장됩니다 | |
| Output | Folder | output_dir | path/to/output_dir | 게놈 인덱스가 저장되는 디렉토리(이하 "게놈 디렉토리"라고 함)로의 경로를 지정합니다. 이 디렉토리는 STAR 실행 전에 (mkdir로) 생성되어야 하며, 쓰기 권한이 있어야 합니다. 파일 시스템에는 일반적인 포유류 게놈에 사용할 수 있는 최소 100GB의 디스크 공간이 필요합니다. 게놈 생성 단계를 실행하기 전에 모든 파일을 게놈 디렉토리에서 제거하는 것이 좋습니다. 이 디렉토리 경로는 참조 게놈을 식별하기 위해 매핑 단계에서 제공되어야 합니다. |
결과
-
.png)
genomeParameters.txt는 해당 STAR 인덱스를 만들 때 사용된 모든 생성 옵션과 파생 값(STAR 버전, 참조 FASTA·GTF 경로, sjdbOverhang, genomeSAindexNbases, 크기/메모리 관련 수치 등)을 기록한 메타데이터야. 이 파일로 인덱스의 재현성·호환성(예: 리드 길이와 sjdbOverhang 일치, 버전 확인)을 빠르게 점검하고, 동일 조건으로 다시 인덱스를 만들 때 참고할 수 있다.
STAR2
STAR2는 "Spliced Transcript Alignment to a Reference Genome"의 약자로, NGS 데이터를 참조 게놈에 정렬하는 도구로 사용됩니다. 주로 RNA-Seq 데이터를 처리하기 위한 고성능의 시퀸스 정렬 도구 중 하나입니다. STAR2는 STAR의 두 번째 버전으로 업데이트되었으며, STAR에 비해 더 빠르고 효율적인 정렬 기능과 다양한 기능을 제공합니다.
STAR2는 다른 도구에 비해 긴 리드의 정렬도 정확하고 효과적으로 처리할 수 있어 다양한 시퀸싱 플랫폼에서 얻은 데이터를 다룰 수 있습니다. 입력 데이터로 사용하는 파일은 fastq, fastq.gz, fq, fq.gz 확장자를 가진 파일입니다. 또한, genome 상의 각 위치에 대한 정보를 담고 있어 정렬 속도를 높이고 정확한 매핑을 가능케 하는 genome index 파일과 genome의 실제 시퀸스 정보를 포함하고 있는 Reference 파일도 필요합니다.
출력으로는 정렬이 완료된 BAM 파일(또는 sam 파일)과 매핑 작업이 완료된 후에 나오는 요약 매핑 통계 파일(final.out), 실행에 대한 자세한 정보가 담긴 로그 파일(Log.out), 처리된 read의 수 등 작업 진행 통계를 보고하는 파일(progress.out), 각 splice에 대한 정보가 담긴 정보 파일(out.tab)까지 총 5가지의 파일이 생성됩니다.
주요사항
- STAR2(STAR)는 RNA-seq 리드를 유전체에 초고속 정렬하고 스플라이스 접합을 탐지하는 도구로, 사용 전 genome.fa+genes.gtf로 **인덱스(star2_index)**를 만들어 둬야 한다. --sjdbGTFfile(주석 반영), --sjdbOverhang=읽기길이-1(스플라이스 감도), --runThreadN(스레드), 필요 시 —genomeSAindexNbases·메모리 한도. 버전/참조·주석/리드 길이가 바뀌면 인덱스 재생성이 안전하며, 출력은 정렬된 BAM, 접합 목록(SJ), 로그·QC 통계가 기본이다.
입력 데이터 예제
실행 명령어 예시
./STAR2.sh \ input_dir = "./cutadapt/output" \ star_index_dir = "./star2_index/output" \ output_dir = "./star2/output" \ is_paired = "TRUE"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./cutadapt/output | 매핑할 시퀀스가 포함된 파일(예: RNA-seq FASTQ 파일)을 포함하는 디렉토리 경로. 일루미나 페어드 엔드 읽기를 사용하는 경우 read1 및 read2 파일을 제공해야 합니다. | |
| Input | Folder | star_index_dir | ./star2_index/output | 생성된 유전체 인덱스가 있는 유전체 디렉토리의 경로를 지정합니다 | |
| Output | Folder | output_dir | ./star2/output | Star2를 실행한 후 결과가 저장될 디렉토리 경로 | |
| Option | Boolean | is_paired | TRUE | 단일 끝 또는 희미한 끝 중 하나를 선택합니다. 기본값은 "true"입니다 |
결과
-
.png)
Log.out은 STAR가 무엇을 어떤 옵션으로 실행했는지 전 과정을 기록한 실행 로그이다. 주요 내용은 STAR 버전·실제 실행된 command line, 입력 파일/인덱스 경로, 재정의된 파라미터(sjdbOverhang, genomeSAindexNbases 등), 인덱스 생성 시 genomeParameters, 경고/에러 메시지 등이다.
-
.png)
Log.progress.out은 STAR 정렬이 진행되는 동안 실시간 누적 통계를 찍는 파일이다. 시간대별로 처리 속도(M/hr), 누적 read 수·길이, Mapped unique/ multi(%), MMrate(불일치율), Unmapped(short/other %) 등을 보여줘서, 실행 중 성능과 매핑률 추이를 모니터링하고 이상 징후(급격한 매핑률 저하 등)를 즉시 파악할 수 있다. 마지막 줄의 **ALL DONE!**은 정렬 완료를 뜻한다.
-
.png)
Log.final.out은 STAR 정렬의 최종 품질 요약표로, 전체 읽기 수·평균 길이, Uniquely mapped %, Multi-mapping %, 스플라이스(Annotated/Non-canonical), 불일치/삽입·삭제율, Unmapped 사유(too short/too many loci 등)와 처리 속도를 한 번에 보여준다. 보고서엔 보통 Uniquely mapped ≥ ~70–80%, MMrate(불일치율) 낮음, Unmapped too short 낮음 등을 확인해 라이브러리 품질과 정렬 성능을 평가한다.
-
.png)
STAR의 SJ.out.tab(splice junction 목록)이다. 각 행이 하나의 접합을 뜻하고, 열은 아래와 같다: 염색체, 2) 인트론 시작(1-based), 3) 인트론 끝, 4) 가닥(0=미지정, 1=+, 2=−), 5) 모티프(0=비정형, 1=GT/AG, 2=CT/AC, 3=GC/AG, 4=CT/GC, 5=AT/AC, 6=GT/AT), 6) 주석여부(0/1), 7) 유니크 매핑 read 수, 8) 멀티 매핑 read 수, 9) 최대 스플라이스 오버행. 예) 1 14475 15854 2 4 0 0 1 37 → chr1의 [14475–15854] 인트론, 음성 가닥(2), 모티프 CT/GC(4), 주석 없음(0), 멀티매핑 1건, 최대 오버행 37bp.
featurecounts
featurecounts는 주어진 정렬된 BAM 또는 SAM 파일에서 각 유전자 또는 feature에 할당된 리드 수를 계산합니다. featurecounts는 각 feature에 할당된 리드 수를 담은 count matrix를 출력하여 발현 수준을 비교하거나 Downstream 분석을 위한 입력 데이터로 활용할 수 있습니다. 입력 데이터로는 여러 개의 SAM 또는 BAM 파일이 사용되며, 출력 데이터로는 각 유전자에 할당된 리드 수를 담은 count matrix가 생성됩니다.
※ count matrix 파일
RNA 시퀀싱 분석에서 얻은 유전자 발현량을 정리한 행렬 형태의 데이터 파일입니다. 여러 샘플에서 각 유전자가 얼마나 발현되었는지를 나타내는 정량적 수치를 담고 있습니다. 특히 featureCounts 나 htseq-count와 같은 도구를 통해 생성된 원본 read count 값을 사용하며, 이는 정규화되지 않은 상태의 데이터입니다.
주요사항
- featureCounts는 정렬된 BAM/CRAM에서 유전자별 read 카운트 매트릭스를 만드는 빠른 카운터다(DESeq2/edgeR 입력). 표준 사용: -a genes.gtf -t exon -g gene_id(+ -T 스레드, -s 방향성, -p -B -C 페어드 처리, 멀티매핑은 -M/--fraction 선택). 출력은 counts.txt와 summary(Assigned/Unassigned 사유)로, 이 요약으로 라이브러리 품질·설정 적절성(Directional, MAPQ 등)을 점검한다.
입력 데이터 예제
실행 명령어 예시
Rscript featureCounts.r input_dir = "./star2/output" \ output_dir = "./featureCounts/output"\ annotation = "hg38" \ is_paired_end = "TRUE"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./star2/output | 읽기 매핑 결과가 포함된 입력 파일의 이름을 나타내는 문자 벡터. 파일은 SAM 형식 또는 BAM 형식일 수 있습니다. 파일 형식은 함수에 의해 자동으로 감지됩니다. | |
| Output | Folder | output_dir | ./featureCounts/output | 분석 도구를 실행한 후 결과가 저장될 디렉토리 경로 | |
| Option | String | annotation | hg38 | 읽기 요약에 사용되는 내장 주석을 지정하는 문자열입니다. 이 문자열은 각각 유전체 'mm10', 'mm9', 'hg38' 및 'hg19'에 대한 NCBI RefSeq 주석에 해당하는 mm10, mm9, hg38 및 hg19를 포함한 네 가지 가능한 값을 가지고 있습니다. 기본적으로 mm10입니다. 내장 주석은 SAF 형식을 가지고 있습니다(아래 참조). | |
| Option | Boolean | is_paired_end | False | 논리적으로 페어드 엔드 읽기가 사용되는지 여부를 나타냅니다. TRUE인 경우 개별 읽기 대신 조각(템플릿 또는 읽기 쌍)이 계산됩니다. 기본적으로 FALSE입니다. |
결과
-
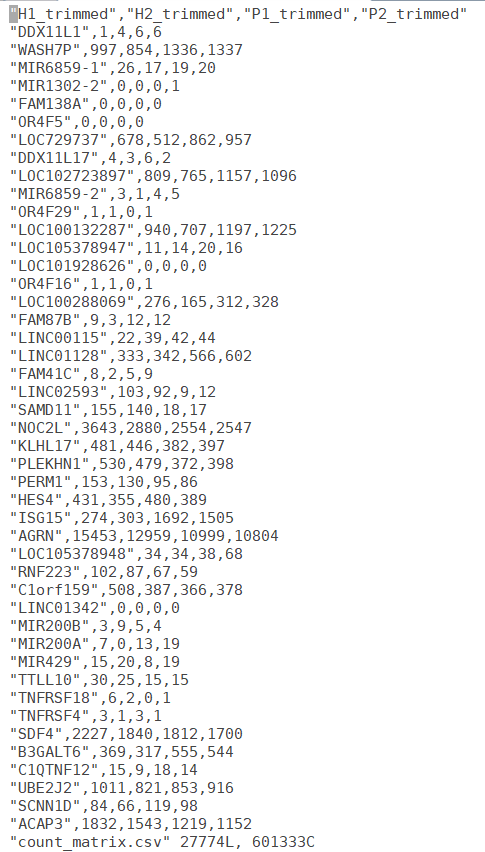
RNA 시퀀싱 분석에서 얻은 유전자 발현량을 정리한 행렬 형태의 데이터 파일입니다. 여러 샘플에서 각 유전자가 얼마나 발현되었는지를 나타내는 정량적 수치를 담고 있습니다. 특히 featureCounts 나 htseq-count와 같은 도구를 통해 생성된 원본 read count 값을 사용하며, 이는 정규화되지 않은 상태의 데이터입니다.
TPMcalculator
TPMCalculator는 주어진 정렬된 BAM 또는 SAM 파일에서 각 샘플마다 유전자에 할당된 리드 수를 기반으로 유전자 길이를 고려하여 보정하여 정규화한 값을 계산합니다. TPM (Transcripts per million) 값은 read count 수를 유전자의 길이로 나눈 값에 103을 곱한 RPK (Reads per kilobase)값을 계산한 후 RPK값을 전체 RPK값으로 나눈 후 106을 곱하여 계산합니다. TPMCalculator는 각 샘플에 할당된 TPM 값을 나타내는 TPM matrix를 출력하여 샘플 간의 유전자 발현 수준을 비교할 수 있습니다. TPMCalculator의 입력 데이터로는 여러 개의 SAM 또는 BAM 파일이 사용되며, 출력 데이터로는 각 유전자에 할당된 TPM값을 나타내는 TPM matrix가 생성됩니다. 입력 데이터로는 RNA-Seq 데이터의 BAM 파일과 유전자 주석 정보를 포함하는 GTF 파일입니다. 출력으로는 TPM 결과 파일과 여러 샘플의 TPM 데이터를 통합한 매트릭스 파일(tpm_matrix.txt)입니다.
주요사항
- TPMCalculator는 좌표정렬 BAM과 GTF/GFF를 입력으로 받아 유전자/트랜스크립트 TPM(옵션에 따라 FPKM/카운트) 테이블을 산출하는 도구이다. STAR/HISAT2 정렬 완료 및 .bai 인덱스, 파이프라인과 동일한 주석 버전, 올바른 방향성(strandedness) 설정이 정확도에 핵심이다. 결과는 샘플 간 발현 정규화 비교와 시각화(히트맵 등)에 바로 쓰기 좋다. 예: TPMCalculator -g genes.gtf -b sample.bam -o out/ -p 8.
입력 데이터 예제
실행 명령어 예시
./TPMCalculator.sh input_dir = "./star2/output" \ gtf_file = "./gtf_file" \ output_dir = "./tpmcalculator/output" \ read_length = "16"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./star2/output | 먼저, BAM 파일이 필요합니다. 이 파일에는 얻은 시퀀싱 읽기의 매핑 결과가 포함되어 있습니다 RNA-seq 실험에서 참조 게놈까지. BAM 파일은 참조 게놈과 대조하여 정렬되어야 합니다, samtools 정렬 명령을 사용하여 처리할 수 있습니다. TPMCalculator를 실행할 때, BAM 파일의 정확한 경로를 지정해야 합니다. | |
| Input | File | gtf_file | ./gtf_file | GTF 또는 GFF 형식의 유전자 주석 파일이 필요합니다. 이 파일에는 위치 정보가 포함되어 있습니다 참조 게놈에 있는 유전자의 구조적 특징. 특히, 여기에는 엑손 위치와 같은 정보가 포함되어 있습니다 전사 시작/종료 사이트, 정확한 표현 수준 계산에 필수적입니다. 이 주석 파일은 일치해야 합니다 RNA-seq 데이터 매핑에 사용되는 참조 게놈 버전, 그리고 TPMCalculator를 실행할 때, GTF 파일의 경로를 지정해야 합니다. | |
| Output | Folder | output_dir | ./tpmcalculator/output | TPMCalculator 결과를 위한 출력 디렉토리 | |
| Option | Integer | read_length | 16 | 더 작은 크기 덕분에 유전자를 위한 인트론이 생성되었습니다. 기본값: 16. 읽기 길이를 사용하는 것이 좋습니다 |
결과
-
.png)
TPM matrix는 행=유전자(Gene_Id), 열=샘플명으로 구성된 발현 매트릭스로, 각 셀 값이 해당 유전자의 TPM(Transcripts Per Million) 정규화 발현량을 뜻한다. 라이브러리 크기와 유전자 길이 보정을 거쳐 샘플 간 비교가 가능한 값이라, 히트맵·클러스터링·마커 선별 등에 바로 사용한다.
DESeq2
eDESeq2는 featurecount를 통해 생성된 전사체 raw count 데이터와 샘플별 실험군과 대조군 분류 양상을 나타내는 design matrix 데이터를 활용하여 실험군과 대조군 사이의 전사체 발현량 차이를 식별하는 데 사용되는 R 패키지입니다. raw count데이터를 Size factor normalization 방법을 사용하여 발현량을 정규화한 후, Negative Binomial Distribution 방법으로 차등 발현 분석을 수행합니다. 이 프로그램은 p값과 log2 fold change 값을 포함한 다양한 통계량이 입력되어 있는 detable.csv파일을 생성합니다,해당 모듈은 우선 입력 데이터로 전사체 count matrix(파일명은 반드시 count_matrix.csv) 파일과 design matrix 파일의 경로를 변수로 입력받습니다. 다음으로 결과 파일이 생성될 디렉토리 경로를 변수로 입력합니다. 마지막 변수로는 design matrix 파일 내 대조군으로 설정한 이름을 입력하여 실행합니다.
※ design matrix 파일
실험 설계와 샘플 그룹 정보를 설명하는 메타데이터 파일입니다. 차등발현분석에 필수적인 정보를 포함하며, 각 행은 샘플에 해당하고 샘플명과 해당 조건(예: control 또는 case)을 포함합니다. 이 파일은 반드시 design_matirx.csv 라는 이름으로 존재해야 하며, count_matrix.csv 파일의 열 이름과 정확히 일치해야 합니다.
주요사항
- raw count_matrix + 설계식(design)을 입력으로 음이항 모델을 사용해 차등발현(DEG)을 검정한다. 라이브러리 보정(크기계수)→ 분산 추정/수축 → log2FC, p-value, padj(FDR)를 산출하며, lfcShrink로 효과 크기 안정화가 가능하다. QC는 vst/rlog 변환 후 PCA·샘플거리·MA-plot을 보고, 실무에선 설계식에 배치(~ condition + batch)를 포함하는 게 핵심이다.
입력 데이터 예제
실행 명령어 예시
Rscript deseq2.r input_dir = "./featurecounts/output" \ design_matrix_file = "./design_matrix_file.csv" \ output_dir = "./deseq2/output" \ control_name = "control"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./featurecounts/output | 분석에 사용될 count matrix 파일이 있는 디렉토리 경로 파일명은 반드시 count_matrix.csv 이어야 함 | |
| Input | File | design_matrix | ./design_matrix_file.csv | 분석에 사용될 design matirx 파일이 있는 디렉토리 경로 파일명은 반드시 design_matrix.csv 이어야 함 | |
| Output | String | output_dir | ./deseq2/output | DESeq2 결과에 대한 출력 디렉토리 | |
| Option | String | control_name | control | 메타데이터 열에 있는 대조군의 이름. |
결과
-

행=유전자, 열=통계지표(baseMean, log2FoldChange, lfcSE, stat, pvalue, padj)로 이뤄진 차등발현 표이다. log2FoldChange는 효과 크기(부호=증가/감소), padj는 다중검정 보정된 유의확률(FDR). 보통 padj < 0.05(또는 0.1)와 |log2FC| ≥ 1로 유의 유전자를 선별하며, 만약 padj가 음수로 보이면 내보내기 중 -log10 변환 여부를 확인한다.
fgsea_enrichment
GSEA_enrichment는 사전에 등록된 특정 기준에 따라 분류한 gene set 들이 DESeq2 분석 결과 유의성을 갖는 유전자들과 얼마나 관련이 있는지를 나타내는 enrichment score와 각종 통계량을 계산하는 R 패키지입니다. fgsea 결과를 enrichment plot으로 나타낼 수 있으며, ggplot 패키지를 활용하여 enrichment bar plot을 출력할 수 있습니다. 추가적으로 pathway별 heatmap 이미지도 생성합니다. GSEA_enrichment는 입력 데이터로는 detable.csv 파일과 count_matrix 파일을 받습니다. 결과물로는 fgsea 결과 CSV 파일이 생성되며, 시각화 자료로는 enrichment plot / enrichment bar plot 그리고 pathway 별 heatmap 이미지가 각각 PNG 파일로 생성됩니다.
주요사항
- 랭크된 유전자 리스트(예: stat/logFC)와 gene set(GMT)을 입력으로, 각 경로의 ES/NES, p-value, FDR(padj), leading-edge를 계산해 상·하향 풍부화를 판정한다. 해석은 보통 padj < 0.25(또는 0.05)와 |NES| 크기로 하며, 부호(±)가 활성 방향(상위/하위)을 의미한다. 결과는 리포트(바플롯·러그플롯)와 EnrichmentMap 네트워크로 요약해 군집/핵심 유전자(leading-edge)를 파악하고 후속 생물학적 가설을 제시한다.
입력 데이터 예제
실행 명령어 예시
Rscript fgsea_enrichment.r input_dir = "./deseq2/output" \ count_matrix = "./featurecounts/output" \ output_dir = "./fgesa_enrichment/output" \ stats = "None" \ min_size = "15" \ max_size = "500" \ gsea_param = "1.15" \ gene_set_name = "None" \ bar_plot_sort_parameter = "None" \ kegg_organism = "hsa"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./deseq2/output | 분석에 사용할 detable.csv 파일이 있는 디렉터리 경로 | |
| Input | Folder | count_matrix | ./featurecounts/output | 분석에 사용될 count matrix 파일이 있는 디렉토리 경로 파일명은 반드시 count_matrix.csv 이어야 함 | |
| Output | Folder | output_dir | ./fgesa_enrichment/output | 분석 도구를 실행한 후 결과가 저장될 디렉토리 경로. | |
| Option | String | stats | None | 테이블을 정렬하는 데 사용되는 매개변수 | |
| Option | Integer | min_size | 15 | 분석에 포함될 최소 gene set 크기 | |
| Option | Integer | max_size | 500 | 분석에 포함될 최대 gene set 크기 | |
| Option | Float | gsea_param | 1.15 | GSEA 계산에 사용, 유전자 순위에 따른 가중치 조정 | |
| Option | String | gene_set_name | None | gene set 설정 | |
| Option | String | bar_plot_sort_parameter | None | bar 차트 정렬, ES 또는 NES인 값에 따라 결과가 정렬되고 상위 경로가 추출 | |
| Option | String | kegg_organism | hsa | KEGG_ORGANISM 예: "hsa" (인간), "mmu" (쥐), "rno" (랫드) |
결과
-
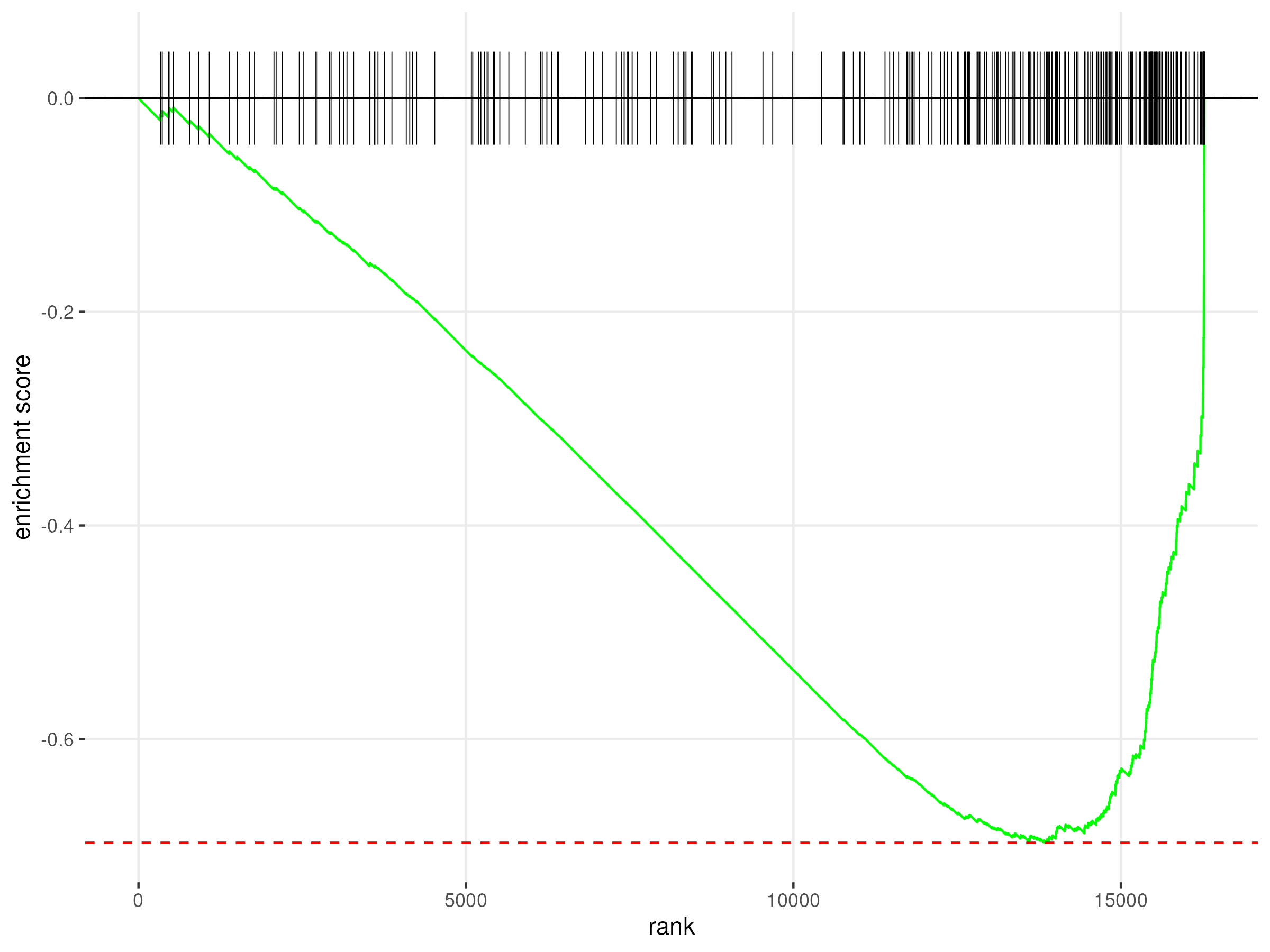
하나의 pathway의 enrichment score 도출 과정을 시각화한 그래프를 생성합니다. fgsea는 각 유전자를 발현량을 기준으로 내림차순으로 정렬합니다. 그래프의 x축에는 검은색 선이 존재하며, 이는 pathway에 속한 유전자들의 발현량 순위를 나타냅니다. 초록색 선은 gene set에 존재하는 유전자들의 발현량을 더하고, 나머지는 빼는 enrichment score 도출 과정을 나타냅니다. 빨간 선은 음수나 양수 중 가장 큰 수를 의미하며, 이중 절대값이 더 큰 수를 enrichment score로 정의합니다.
-
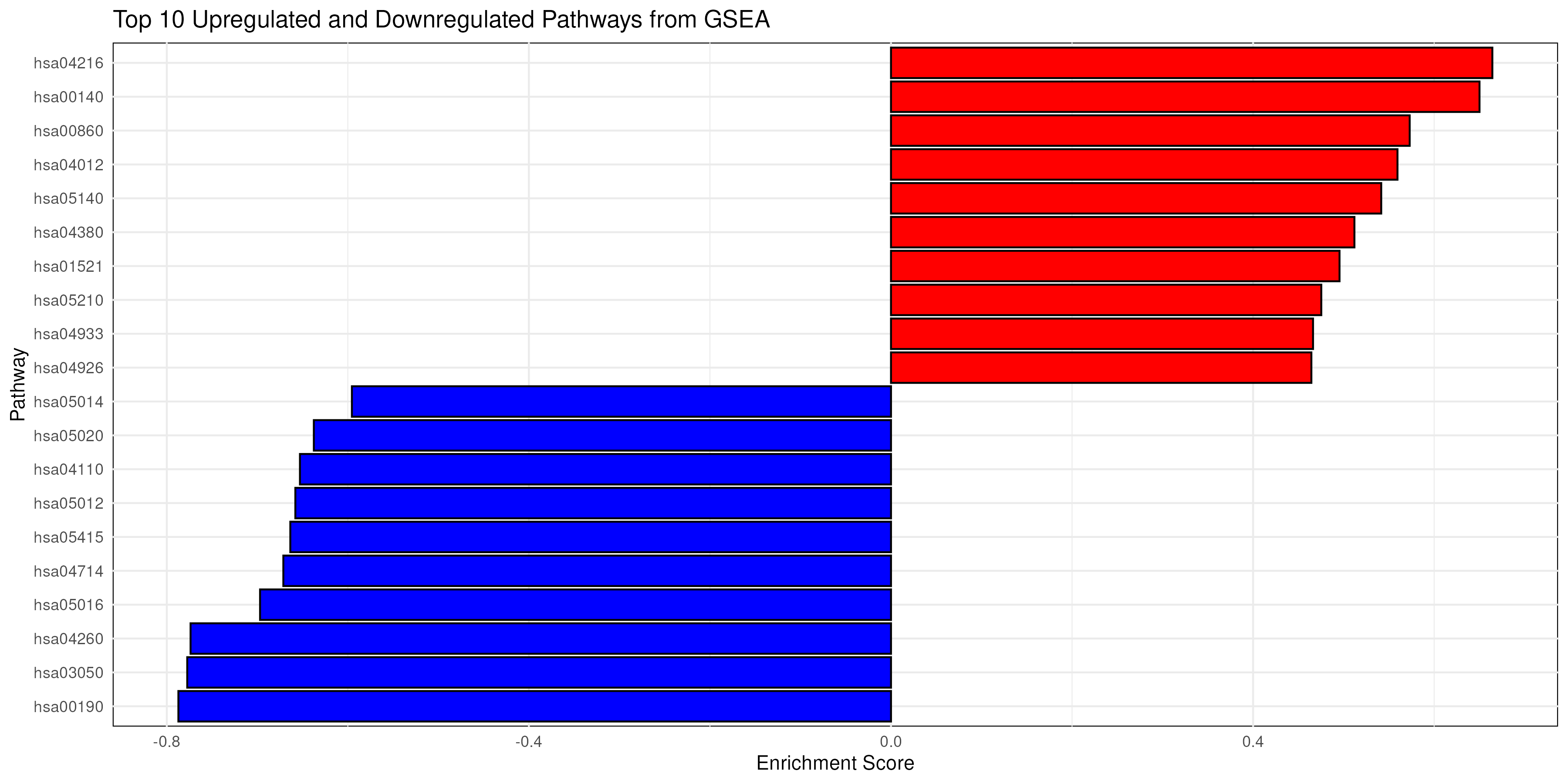
Enrichment score 값을 정규화한 후 상위 10개와 하위 10개의 pathway를 선별하여 막대 그래프를 생성합니다. 그래프의 x축은 정규화된 enrichment score을, y축은 상위 또는 하위 10개의 pathway를 나타냅니다.
-
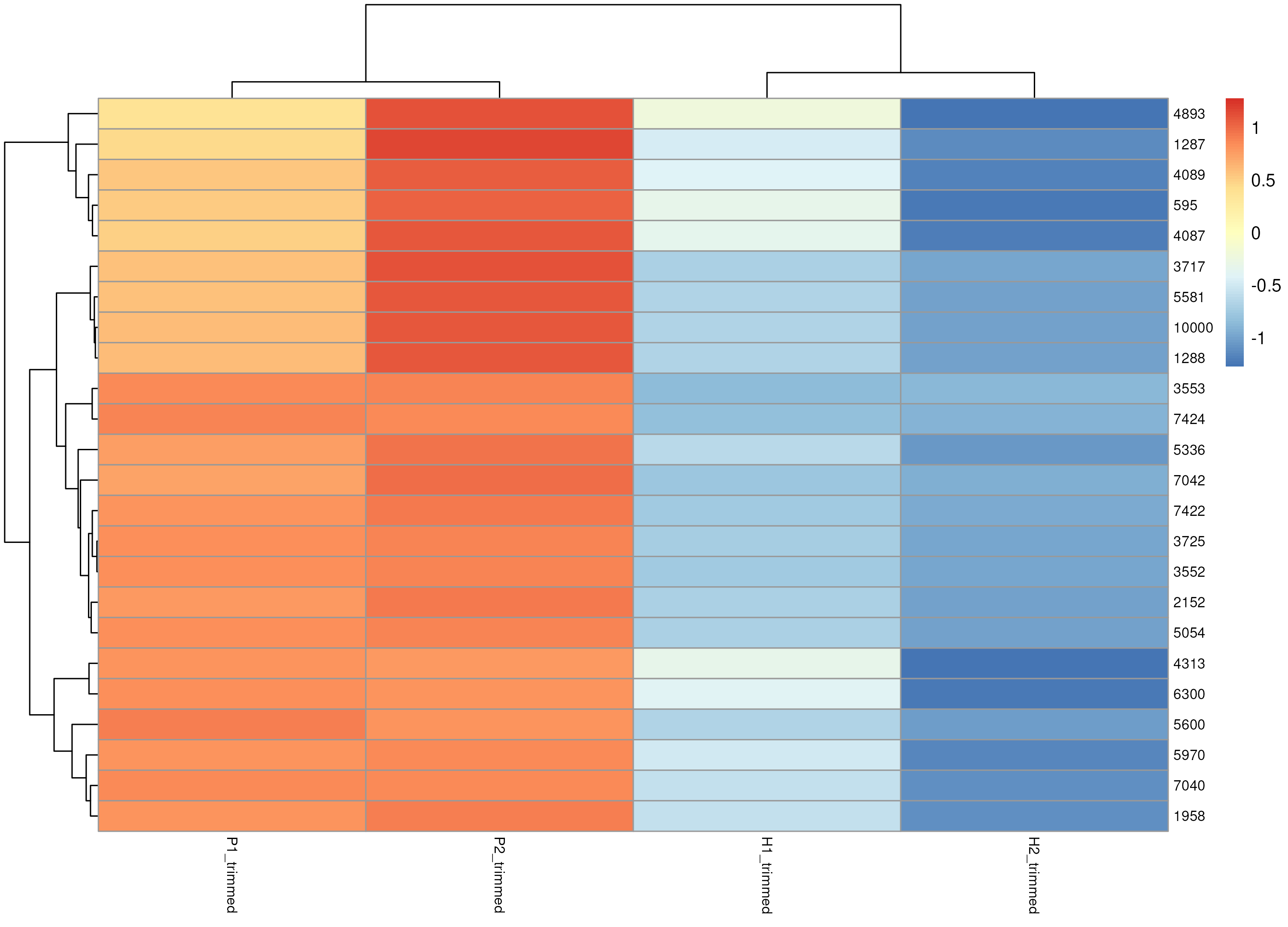
GSEA 풍부화 히트맵으로, 행=경로(geneset), 열=샘플/그룹이며 색은 NES(또는 −log10 FDR) 등 풍부화 강도를 나타냄(빨강=상향, 파랑=하향). 열이 그룹별로 묶여 좌측 집단에서 상향(빨강), 우측 집단에서 하향(파랑)되는 조건 특이적 경로 패턴이 보인다. 같은 색이 연속되는 블록들은 기능 모듈/군집을 시사해, 후속 해석(핵심 경로 선정, 네트워크 요약)에 유용하다.
RNA_Expressionviz
DEG-Viz는 corrleationmap, heatmap, MA plot, Volcano plot과 같은 RNA Expression의 시각화에 사용되는 도구입니다.
■ correlationmap: 높은 발현량의 차이를 보이는 상위 유전자를 추출하여 이들 간의 상관관계를 비교 분석한 표를 출력합니다. DESeq2를 통해 생성된 detable과 count matrix를 입력받아 logFC 값의 절대값이 높은 n개의 유전자 또는 입력받은 유전자를 pearson, spearman 등의 기법을 사용하여 상관관계를 분석합니다. GGally package를 사용하며 출력 파일로는 상관관계 도표 PNG 파일이 생성됩니다.
■ Heatmap: 높은 발현량의 차이를 보이는 상위 n개의 유전자 또는 입력받은 유전자를 추출하여 샘플별로 발현량의 차이를 확인하기 위해 heatmap을 그립니다. Pheatmap 패키지를 사용하며 출력파일로는 heatmap PNG 파일이 생성됩니다.
■ MA plot: 각 유전자에 대한 logCPM과 logFC 항목을 활용하여 산포도를 그립니다. ggplot2 패키지를 사용하며, MA plot PNG 파일을 생성합니다.
■ Volcano plot: 보정된 p값과 logFC 항목을 사용하여 실험군과 대조군 샘플 사이의 발현량 차이와 유의성에 따른 분포를 확인할 수 있는 volcano plot을 그립니다. ggplot2 패키지를 사용하여 volcano plot PNG 파일을 생성합니다.
주요사항
- RNA-seq 발현 데이터를 시각화하는 도구로, TPM/FPKM 행렬 또는 DESeq2 결과(정규화 카운트·VST, DEG 테이블)를 입력으로 받아준다. 주요 출력은 히트맵/클러스터링, PCA(또는 UMAP), volcano/MA plot, 박스·바 플롯(마커 유전자)이며, 샘플·유전자 필터링과 스케일링(Z-score) 옵션을 지원한다. 실무 팁: 입력과 메타데이터(조건/배치) 열 이름 일치, 로그/정규화 상태 확인(VST 권장), 그리고 집단 색상 팔레트·주석 바를 맞춰 레포트용 그림을 일관되게 만든다.
입력 데이터 예제
실행 명령어 예시
Rscript rna_expressionviz.r input_dir = "./deseq2/output" \ count_matrix = "./featurecounts/output" \ output_dir= "./rna_expressionviz/output" \ correlation_genes = "10" \ method = "pearson" \ heatmap_genes = "100" \ row_clustering = "euclidean" \ col_clustering = "euclidean" \ scale = "row" \ dot_size = "1" \ p_value = "0.5" \ fc = "0.5"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./deseq2/input | 분석에 사용할 detable.csv 파일이 있는 디렉터리 경로 | |
| Input | Folder | count_matrix | ./featurecounts/output | 분석에 사용될 count matrix 파일이 있는 디렉토리 경로 파일명은 반드시 count_matrix.csv 이어야 함 | |
| Output | String | images | ./rna_expressionviz/output | RNA-expressionViz를 실행한 후 결과가 저장될 디렉토리 경로 | |
| Option | String | correlation_genes | 10 | 상관 분석에 사용할 특정 유전자 목록(콤마로 구분). 입력하면 correlation_top_genes_count가 사용되지 않습니다. | |
| Option | String | method | pearson | 상관관계 계산 방법(피어슨, 스피어맨, 켄달) | |
| Option | String | heatmap_genes | 100 | "히트맵 시각화에 사용할 특정 유전자 목록(콤마로 구분). 입력하면 correlation_top_genes_count가 사용되지 않습니다." | |
| Option | String | row_clustering | euclidean | "히트맵에서 행(유전자) 클러스터링을 위한 거리 계산 방법 예) ""euclidean"", ""manhattan"", ""maximum"", ""canberra"", ""binary"", ""minkowski""" | |
| Option | String | col_clustering | euclidean | "히트맵에서 행(유전자) 클러스터링을 위한 거리 계산 방법 예) ""euclidean"", ""manhattan"", ""maximum"", ""canberra"", ""binary"", ""minkowski""" | |
| Option | String | scale | row | "히트맵 데이터의 스케일링 방법(행, 열 또는 없음) 예) ""row"", ""column"", ""none""" | |
| Option | Integer | dot_size | 1 | MA 플롯과 화산 플롯의 점 크기 | |
| Option | Float | p_value | 0.5 | 유의미한 미분 표현식을 결정하기 위한 P-값 임계값 | |
| Option | Float | fc | 0.5 | 유의미한 미분 표현을 결정하기 위한 접힘 변경 임계값 |
결과
-
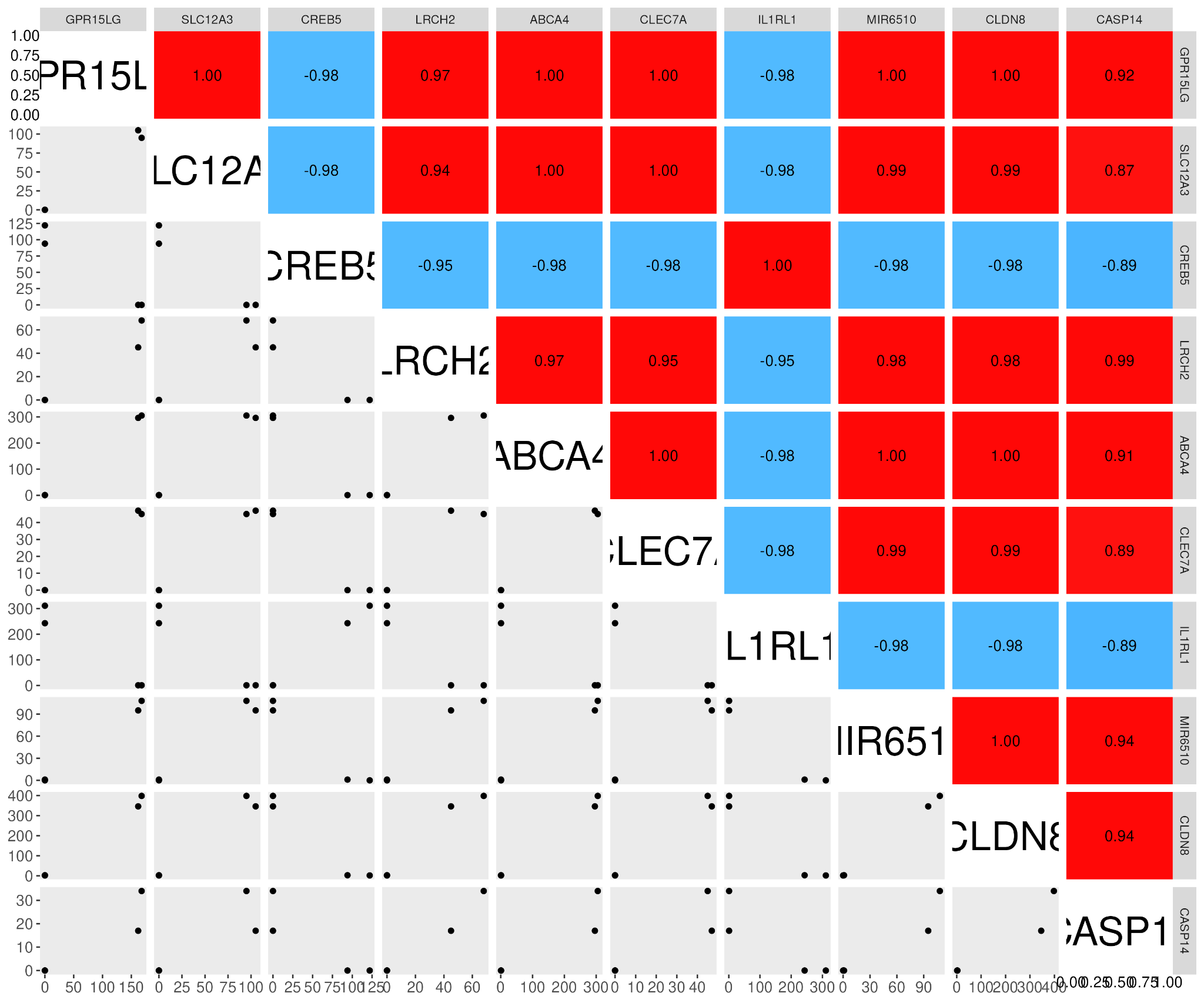
다음은 상위 10개의 유전자 간 상관관계를 나타내는 표를 생성하는 작업입니다. 이 작업에서는 가장 높은 발현량을 보인 상위 10개의 유전자가 선택되어 사용됩니다. 유전자 간의 양의 상관관계는 숫자가 1에 가까우며, 이러한 관계는 상자가 붉은색을 띠게 시각적으로 표현됩니다. 반면에 유전자 간의 음의 상관관계는 숫자가 -1에 가깝게 되며, 이러한 관계는 상자가 푸른색을 띠게 시각적으로 표현됩니다. 이러한 방식으로 상위 10개의 유전자 간의 상관관계를 시각적으로 파악할 수 있습니다.
-
.png)
다음 작업은 각 샘플별로 유전자의 발현량을 평균 내어 정규화한 플롯을 생성하는 것입니다. 플롯에서 푸른색은 해당 유전자의 발현량이 평균에 미치지 못하는 것을 나타내며, 붉은색은 평균을 상회하는 것을 의미합니다. 이러한 방식으로 유전자의 발현량이 유사한 유전자들끼리 묶는 Euclidean clustering 기법을 활용하여 데이터를 시각적으로 분석할 수 있습니다.
-
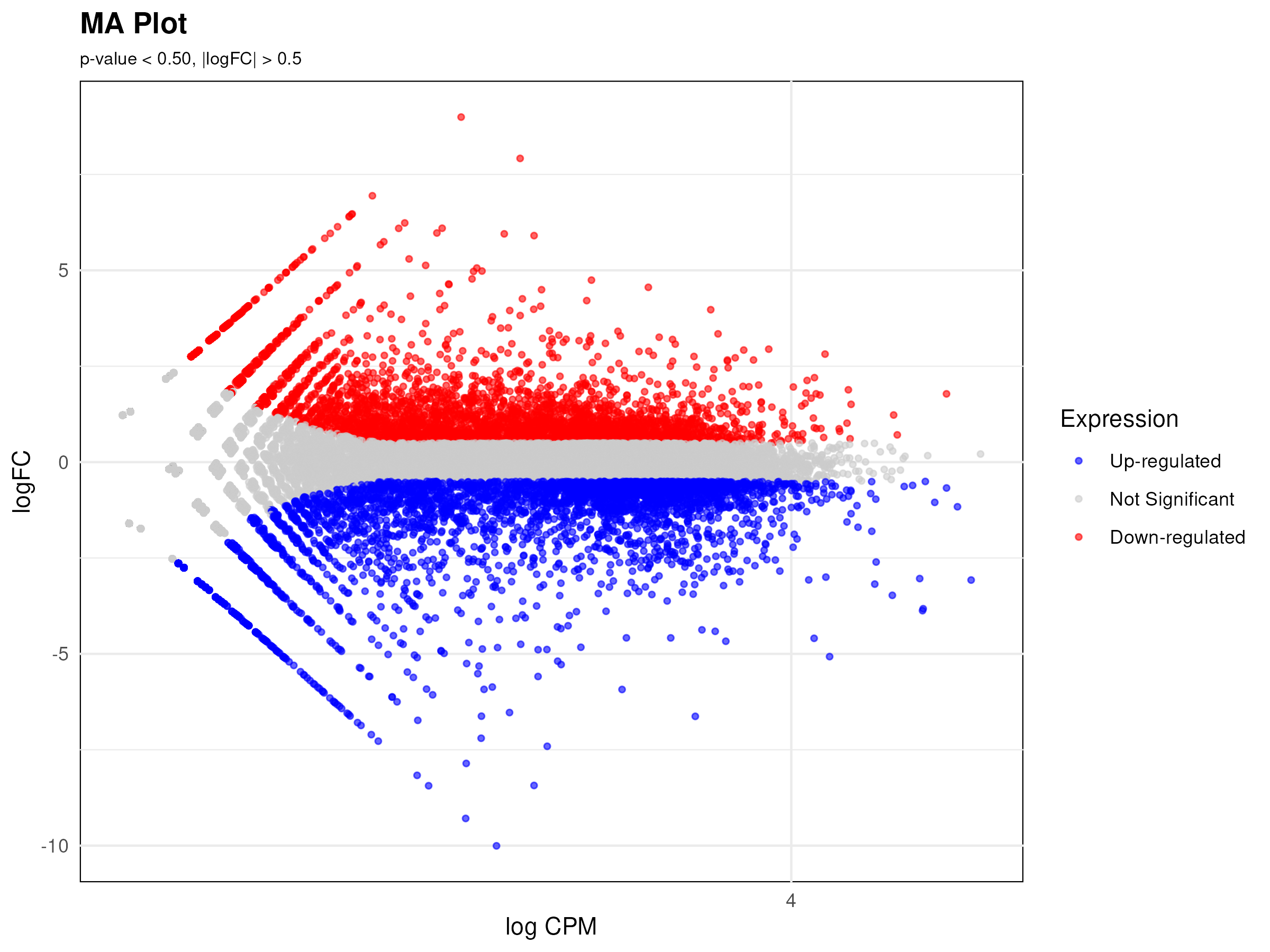
다음 작업은 각 유전자들이 2개의 항목에 대해 보이는 분포를 시각화하는 그래프를 생성하는 것입니다. 여기서 x축은 logCPM을, y축은 log2fold change을 나타냅니다. CPM은 각 샘플별로 read 수를 백만 단위로 정규한 발현량을 나타내며, Fold change는 대조군과 실험군 사이의 발현량의 비를 의미합니다. 이 그래프를 통해 유전자들 간의 발현 패턴을 비교하고 분석할 수 있습니다.
-
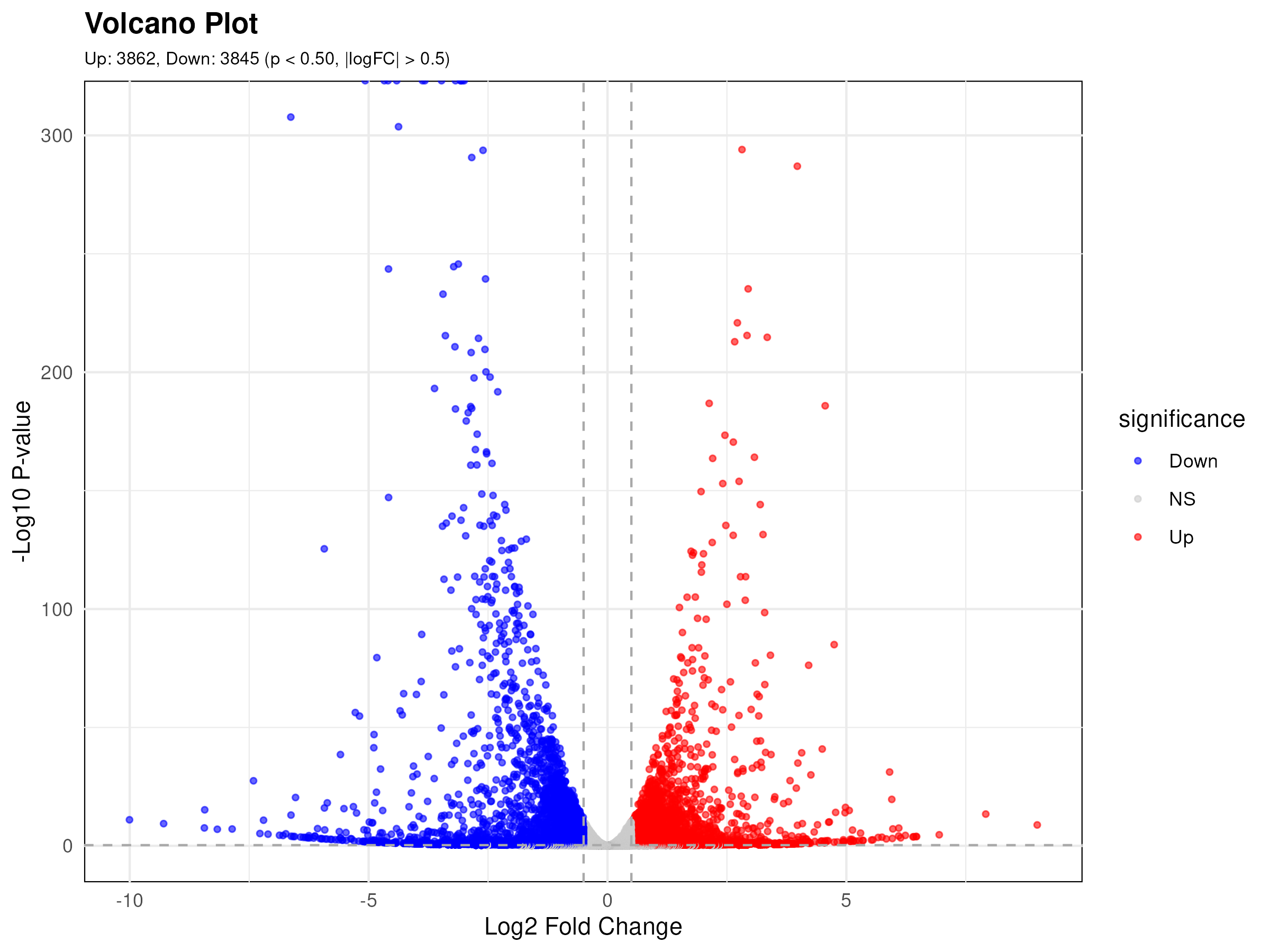
Volcano plot은 실험군과 대조군 간의 유전자 발현 차이를 시각적으로 나타내는 그래프로, 특정 유전자의 발현량 변화와 그 통계적 유의성을 동시에 파악할 수 있습니다. 이 그래프에서 X축은 유전자 발현량 비율의 log2 값으로, 발현량의 크기를 나타냅니다. Y축은 p값의 –log10 값으로, 유전자 발현량 변화의 통계적 유의성을 표현합니다.
fgsea_network
GSEA_network는 fgsea 결과를 바탕으로 gene set 사이의 공통된 유전자들이 얼마나 있는지를 수치화하여 이를 바탕으로 네트워크를 구성합니다. 네트워크를 구축하기 위해 사용된 수치는 jaccard similarity로 gene set 사이의 교집합을 합집합으로 나누어 수치화합니다. 네트워크에 노드로 표시할 gene set은 p값과 보정된 p값 그리고 jaccard similarity를 바탕으로 필터링합니다. GSEA_network는 fgsea를 수행하여 얻은 결과를 바탕으로 ggplot2와 ggraph 패키지를 사용하여 네트워크 PNG 파일을 생성합니다.
주요사항
- GSEA/fgsea 결과를 네트워크(EnrichmentMap)로 시각화해 노드=경로, 엣지=겹치는 유전자(유사도)를 보여주는 분석이야. 노드 색/크기로 방향(NES ±)·강도(|NES|, FDR)를 표현하고, 엣지는 Jaccard/Overlap 기준으로 연결해 군집(모듈)과 허브 경로를 한눈에 파악한다. 실무 팁: 입력을 padj<0.25(또는 0.05)와 |NES|로 필터하고 엣지 유사도 컷오프를 설정해 노이즈를 줄인 뒤, 클러스터 자동 라벨링(AutoAnnotate)로 요약하면 좋다.
입력 데이터 예제
실행 명령어 예시
Rscript fgsea_network.r input_dir = "./deseq2/output" \ output_dir = "./fgsea_network/output" \ stats = "rank_stat" \ min_size = "2" \ max_size = "500" \ gsea_param = "1.8" \ gene_set_extraction_p_value = "0.10" \ gene_set_extraction_adj_p_value = "1" \ jaccard_threshold = "0.10" \ min_overlap_genes = "2" \ overlap_coef_threshold = "0.20" \ topk_when_few = "12"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./deseq2/output | 분석에 사용할 detable.csv 파일이 있는 디렉터리 경로 | |
| Output | String | output_dir | ./fgsea_network/output | 분석 도구를 실행한 후 결과가 저장될 디렉토리 경로. | |
| Option | String | stats | rank_stat | 테이블을 정렬하는 데 사용되는 매개변수 | |
| Option | Integer | min_size | 2 | 최소 유전자 세트 크기 | |
| Option | Integer | max_size | 500 | 최대 유전자 세트 크기 | |
| Option | Integer | gsea_param | 1.8 | 이상치를 제어하기 위해 STAT_1 값을 증가시키거나 감소시키는 Gsea 매개변수 | |
| Option | Float | gene_set_extraction_p_value | 0.10 | 유전자 집합을 추출하기 위한 P-값 매개변수 | |
| Option | Float | gene_set_extraction_adj_p_value | 1 | 유전자 세트를 추출하기 위해 조정된 p-값 매개변수 | |
| Option | Float | jaccard_threshold | 0.10 | 두 경로 A,B의 Jaccard 지수 |A∩B| / |A∪B|가 이 값 이상일 때만 엣지 생성 | |
| Option | Integer | min_overlap_genes | 2 | 엣지를 만들 최소 겹치는 유전자 수 |A∩B| 하한 | |
| Option | Float | overlap_coef_threshold | 0.20 | |A∩B| / min(|A|,|B|) 하한 | |
| Option | Integer | topk_when_few | 12 | padj/|NES| 필터 통과 경로가 적을 때 상위 K개를 추가 표시해 네트워크가 비지 않도록 보장. |
결과
-
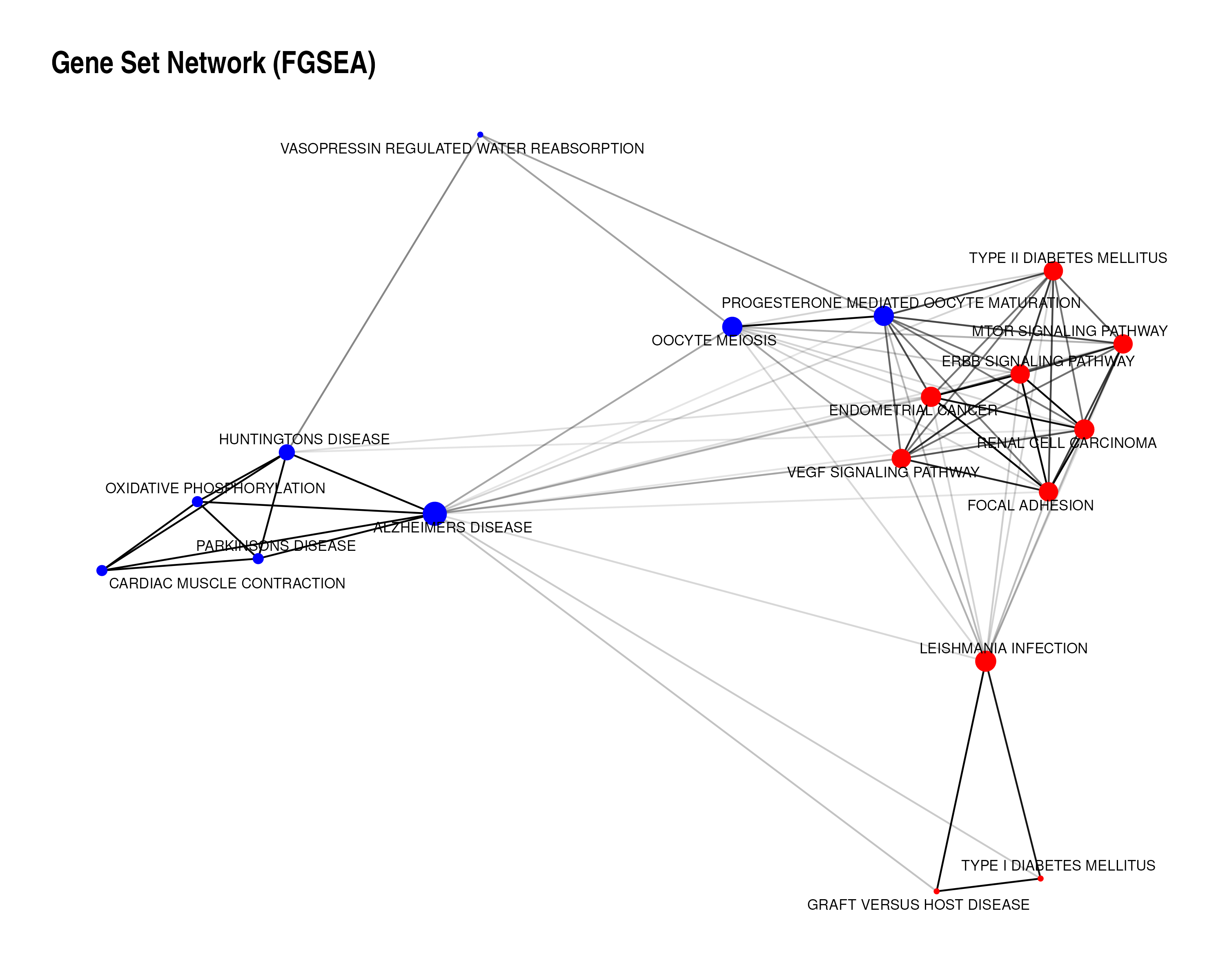
겹치는 유전자의 존재 여부를 나타내는 그래프를 생성합니다. 이 그래프는 fgsea 결과 중 p값이 0.005보다 작거나, 보정된 p값이 0.05 미만인 gene set만을 추출하여 구성됩니다. 두 gene set 간의 관계 정도는 Jaccard similarity 계수를 사용하여 나타냅니다.
Gene_network
이 모듈은 림마 및 엣지R 패키지를 사용하여 디테이블의 출력을 활용합니다. p-값이 0.05 이하로 조정된 유전자를 추출하여 상위 100개의 선택된 유전자를 매핑합니다. stringdb에서 고유 식별자로 이동하여 네트워크를 생성합니다.
주요사항
- 유전자 간 상호작용/공발현/경로-겹침을 노드(유전자·경로)와 엣지(상관·PPI·공유유전자 등)로 시각화·분석해 모듈(클러스터)과 허브 유전자를 찾는 도구이다. 입력은 **발현행렬/DEG 리스트/경로 결과(GSEA·fgsea)**이며, 엣지 가중치는 상관계수·PPI 신뢰도·Jaccard/Overlap처럼 선택해 만들고, 레이아웃/커뮤니티 탐지로 요약한다. 해석 팁: 허브·브리지 노드(degree/betweenness↑)와 모듈의 기능 풍부화를 함께 보고, 다중검정·배치효과·샘플 수에 따른 위양성을 주의한다.
실행 명령어 예시
DEGs <- subset(res, adj.P.Val < 0.05) string_db <- STRINGdb$new(version = "11.5", species = 9606, protocol="http") genes_mapped <- string_db$map(DEG_proteins_df, "Gene",removeUnmappedRows = TRUE)
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | /path/to/input_dir | 분석에 사용할 detable.csv 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | /path/to/output_dir | 분석 도구를 실행한 후 결과가 저장될 디렉토리 경로. | |
| Option | Float | deg_p_value | 0.05 | 조정된 p-값을 기반으로 네트워크를 그리는 유전자 | |
| Option | String | species | 9606 | 분석할 종들 | |
| Option | Boolean | remove_unmapped_rows | TRUE | 연결되지 않은 행 제거 |
결과
-
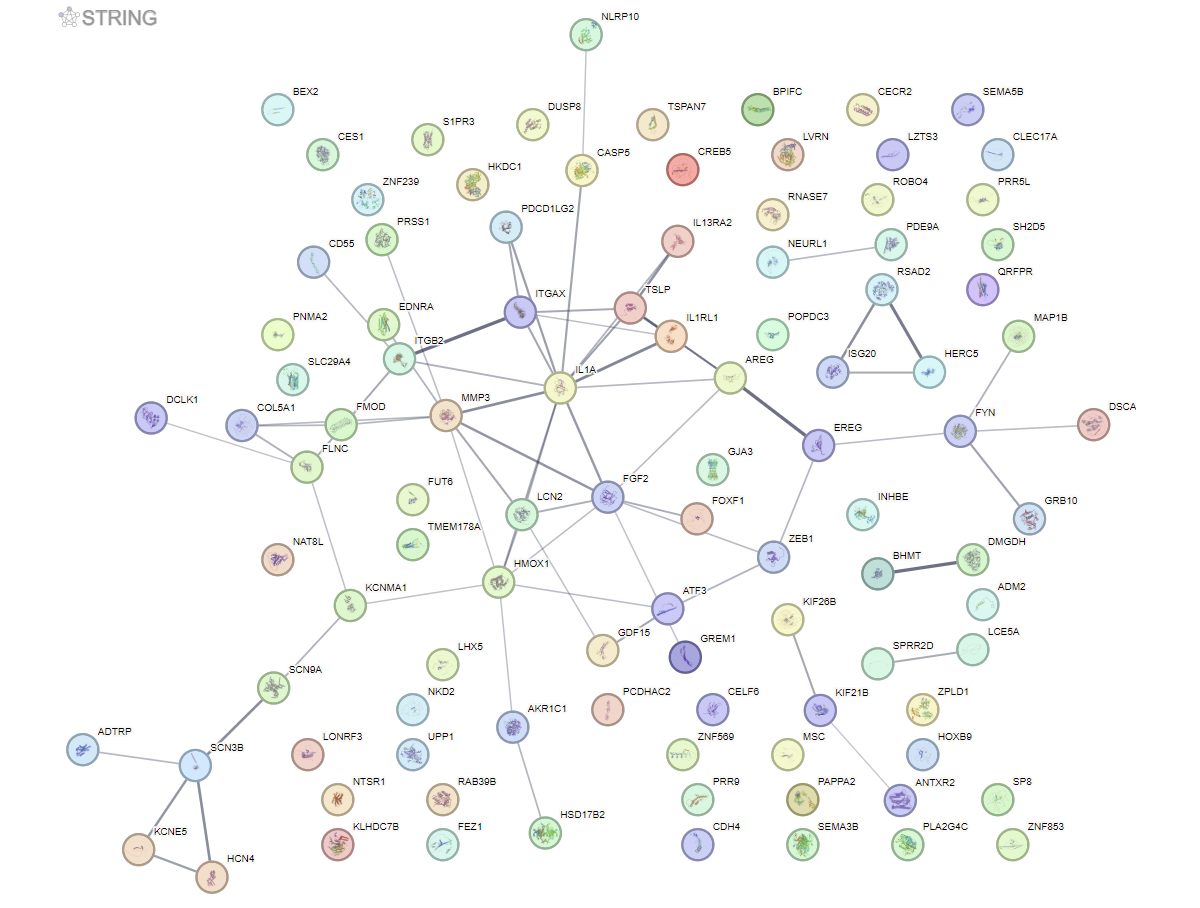
다음 작업은 각 유전자들 간의 관계를 시각화하는 그래프를 생성하는 것입니다. 각 점은 노드로 표시되며, 각 유전자를 나타냅니다. 노드 사이의 선은 유전자들 간의 관계를 나타냅니다. 이 그래프에서는 설정한 pvalue 이하인 유전자들만을 추출하여 시각화합니다. 이를 통해 유전자들 간의 상호 작용과 연관성을 파악할 수 있습니다.
Cluster_profiler
Cluster_profiler는 DESeq2 패키지를 사용하여 출력된 detable 결과 중 보정된 p값과 logFC 항목을 사용하여 실험군과 대조군 샘플 사이의 발현량 차이가 큰 유전자들의 생물학적 과정과 관련한 기능들을 분석한 결과를 나타냅니다. Cluster_profiler는 clusterProfiler 패키지와 ggplot2 패키지를 사용하여 GO_barplot.png파일을 생성합니다.
주요사항
- R에서 과대표현(ORA)·순위기반 GSEA를 수행하는 패키지로, enrichGO/KEGG, gseGO/KEGG, compareCluster 등으로 단일·다군 비교를 지원한다. 입력은 유전자 ID + 종(DB)이며 org.Hs.eg.db/msigdbr/GMT를 쓰고, keyType·universe·pAdjustMethod(FDR) 설정이 정확도와 해석에 핵심이다. 시각화는 dotplot, emapplot(네트워크), cnetplot, ridgeplot 등이 대표적이며, setReadable()로 ID→gene symbol 변환해 읽기 좋게 만든다.
입력 데이터 예제
실행 명령어 예시
Rscript cluster_profiler.r input_dir = "./deseq2/output" \ output_dir = "./cluster_profiler/output" \ gene_counts = "11" \ up_down = "up"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./deseq2/output | 분석에 사용할 detable.csv 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./cluster_profiler/output | 분석 도구를 실행한 후 결과가 저장될 디렉토리 경로. | |
| Option | Integer | gene_counts | 11 | Cluster_Profiler 분석에 사용할 차등 발현 유전자의 개수 | |
| Option | String | up_down | up | 실험군에서 Up-regulation(up)된 유전자인지 Down-regulation(down)된 유전자인지 여부 |
결과
-
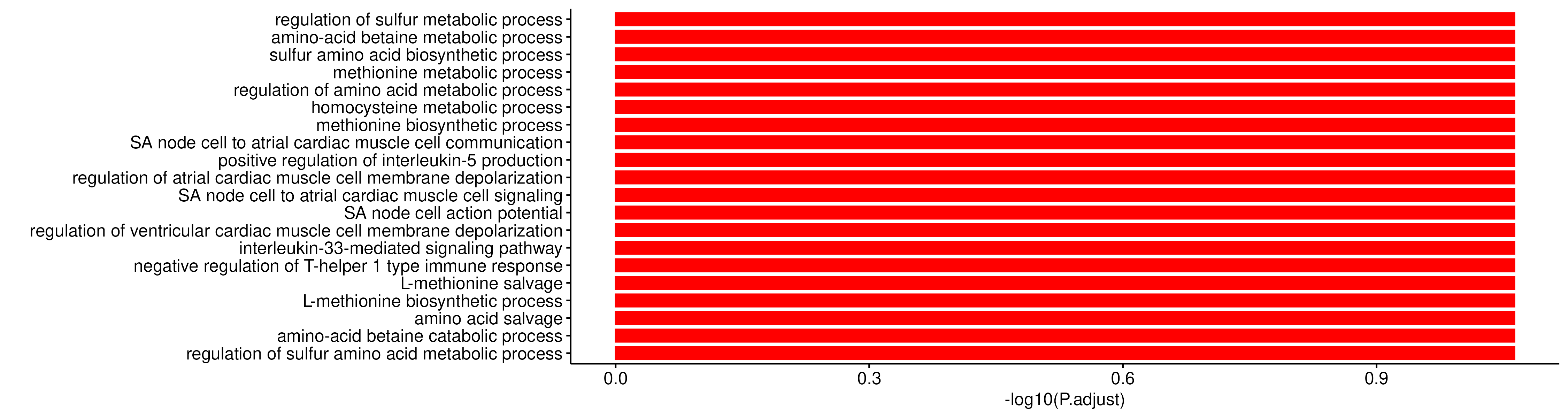
GO_barplot.png은 실험군과 대조군 간의 발현량에 차이가 나는 유전자들의 생물학적 과정과 관련한 기능들을 시각적으로 나타내는 그래프로, 차등 발현 유전자의 생물학적 기능과 그 통계적 유의성을 동시에 파악할 수 있습니다. 이 그래프에서 X축은 adjusted P-value를, Y축은 차등 발현 유전자들의 기능을 나타냅니다.
rmats
Replicate Multivariate Analysis of Transcript Splicing (rMATS)
rMATS turbo는 rMATS의 C/Cython 버전이며(http://rnaseq-mats.sourceforge.net), rMATS과의 주요 차이점은 속도와 저장 공간이다.
rMATS보다 100배 빠르고 출력 파일은 1000배 작은 장점 덕분에 대규모 데이터세트의 분석 및 저장이 쉽고 편리한 도구임.
*MATS는 RNA-Seq 데이터에서 선택적 스플라이싱(AS) 이벤트를 검출하는 계산 도구임.
MATS의 통계적 모델은 두 조건 간 유전자 isoform 비율 차이가 사용자가 정의한 임계값을 초과하는 경우의 P값과 FDR을 계산하며,
RNA-Seq 데이터에서 모든 주요 AS 패턴에 해당하는 이벤트를 자동으로 검출하고 분석할 수 있음.
MATS는 쌍을 이룬 연구 설계와 쌍을 이루지 않은 연구 설계 모두에서 데이터를 처리함.
주요사항
- rMATS는 RNA-seq에서 대체 스플라이싱(AS) 이벤트( SE, A5SS, A3SS, MXE, RI )를 검정하는 도구로, 두 조건 간 ΔPSI(Ψ)·p값/FDR을 산출해 유의한 스플라이싱 변화를 찾는다. 입력은 정렬 BAM(또는 FASTQ+GTF), 시퀀싱 방향성/페어드 정보, read length & libType 등이 필요하며, 복제(rep) 정보를 줘 replicate-aware 통계를 수행한다. 출력은 이벤트별 테이블(각 좌표, 포함/배제 read count, Ψ, ΔΨ, FDR)과 Sashimi 플롯용 파일로, 필터는 보통 |ΔΨ|≥0.1–0.2 & FDR≤0.05를 사용한다.
입력 데이터 예제
실행 명령어 예시
./rmats.sh input_dir = "./star2/output" \ gtf_file = "./gtf_file" \ design_matrix_file = "./design_matrix.csv" \ output_dir = "./rmats/output" \ read_length = "141" \ read_type = "paired" \ lib_type = "fr-firststrand"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./star2/output | 분석에 사용할 detable.csv 파일이 있는 디렉토리 경로 | |
| Input | File | gtf_file | ./gtf_file | GTF 또는 GFF 형식의 유전자 주석 파일이 필요합니다. 이 파일에는 위치 정보가 포함되어 있습니다 참조 게놈에 있는 유전자의 구조적 특징. 특히, 여기에는 엑손 위치와 같은 정보가 포함되어 있습니다 전사 시작/종료 사이트, 정확한 표현 수준 계산에 필수적입니다. 이 주석 파일은 일치해야 합니다 RNA-seq 데이터 매핑에 사용되는 참조 게놈 버전, 그리고 TPMCalculator를 실행할 때, GTF 파일의 경로를 지정해야 합니다. | |
| Input | File | design_matrix_file | ./design_matrix.csv | 분석에 사용될 design matirx 파일이 있는 디렉토리 경로 파일명은 반드시 design_matrix.csv 이어야 함 | |
| Output | String | output_dir | ./rmats/output | 분석 도구를 실행한 후 결과가 저장될 디렉토리 경로. | |
| Option | Integer | read_length | 141 | 각 읽기의 길이. RNA-seq 읽기 길이에 따라 설정된 값을 가진 필수 매개변수. | |
| Option | String | read_type | paired | 분석에 사용된 읽기 유형: 페어드 엔드 데이터의 경우 "페어드" 또는 싱글 엔드 데이터의 경우 "싱글"입니다. | |
| Option | String | lib_type | fr-firststrand | 라이브러리 유형. 스트랜드별 데이터에는 fr-firststrand 또는 fr-secondstrand를 사용합니다. 준비 단계에만 관련이 있고, 사후 단계에는 관련이 없습니다. |
결과
-
.png)
rMATs 실행후 5가지 AS 이벤트 별로 유의미한 결과를 tsv 형식으로 반환함
ggsashimi
ggsashimi는 RNA-seq 데이터를 이용해 대체 스플라이싱(Alternative Splicing) 패턴을 시각화하기 위한 명령줄 기반 도구로, PLoS Computational Biology 저널에 2018년 Diego Garrido-Martin 등이 발표한 오픈소스 소프트웨어다.
주요 특징
- Splicing event 시각화: 지정한 유전체 구간(genomic region)에 대해 스플라이싱 이벤트를 도식화한 Sashimi plot을 생성한다. 개별 샘플뿐 아니라 여러 샘플을 집계(aggregation)하여 평균, 중앙값, 혹은 오버레이 형태로 표시할 수 있다.
- Annotation-independent: 기존 IGV 등의 도구처럼 미리 annotation된 이벤트가 없어도 BAM 파일 정보만으로 splice junction을 추론한다. 즉, 새로 발견된 스플라이싱 이벤트도 탐색 가능하다.
- 표준 포맷 호환성: 표준 정렬 파일(BAM)과 주석 파일(GTF)을 입력으로 사용하며, 결과는 벡터(SVG, PDF) 및 래스터(PNG 등) 이미지로 출력할 수 있다.
주요사항
- 여러 샘플의 RNA-seq 커버리지(엑손)와 스플라이스 접합 아크를 한 그림에 그려주는 파이썬 기반 시각화 도구이다(BAM/BAI + GTF/좌표 입력). 라이브러리 크기 보정(Scaling), Strand/좌표 지정, 샘플 그룹 평균/색상 구분, 접합부에 읽기 수 라벨 표시 등을 지원하며 논문급 그림을 만든다. 사용 예: ggsashimi -b A.bam B.bam -g genes.gtf -r chr1:100000-120000 -M 5 -C 2 -o sashimi.pdf (최소 매핑 접합수·컬러수 등 옵션).
입력 데이터 예제
실행 명령어 예시
./ggsashimi.sh input_dir = "./rmats/output" \ input_bam_dir = "./star2/output_bam" \ gtf_file = "./gtf_file" \ design_matrix_file = "./design_matrix.csv" \ output_dir = "./ggsashimi/output" \ coordinates = "chr2:120225500-120227500" \ alpha = "0.7" \ base_size = "15" \ ann_height = "4" \ width = "6" \ height = "4" \ fdr = "0.05" \ incleveldifference = "0.1" \ event_subtype = "RI,A5SS,A3SS,MXE,SE"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./rmats/output | 분석에 사용할 detable.csv 파일이 있는 디렉토리 경로 | |
| Input | Folder | input_bam_dir | ./star2/output | BAM 파일이 저장된 디렉터리 경로 | |
| Input | File | gtf_file | ./gtf_file | GTF 또는 GFF 형식의 유전자 주석 파일이 필요합니다. 이 파일에는 위치 정보가 포함되어 있습니다 참조 게놈에 있는 유전자의 구조적 특징. 특히, 여기에는 엑손 위치와 같은 정보가 포함되어 있습니다 전사 시작/종료 사이트, 정확한 표현 수준 계산에 필수적입니다. 이 주석 파일은 일치해야 합니다 RNA-seq 데이터 매핑에 사용되는 참조 게놈 버전, 그리고 TPMCalculator를 실행할 때, GTF 파일의 경로를 지정해야 합니다. | |
| Input | File | design_matrix_file | ./design_matrix.csv | 분석에 사용될 design matirx 파일이 있는 디렉토리 경로 파일명은 반드시 design_matrix.csv 이어야 함 | |
| Output | Folder | output_dir | ./ggsashimi/output | 분석 도구를 실행한 후 결과가 저장될 디렉토리 경로 | |
| Option | String | coordinates | chr2:120225500-120227500 | 게놈 영역. 형식: chr:start-end. bam 좌표는 0 기반임을 기억하세요 | |
| Option | Float | alpha | 0.7 | 밀도 히스토그램의 투명도 수준 | |
| Option | Integer | base_size | 15 | 플롯의 기본 글꼴 크기(pch) | |
| Option | Integer | ann_height | 4 | 주석 플롯의 높이(인치) | |
| Option | Integer | width | 6 | 플롯의 너비(인치) | |
| Option | Integer | height | 4 | 개별 신호 플롯의 높이(인치) | |
| Option | Float | fdr | 0.05 | 통계적 유의성 기준 | |
| Option | String | incleveldifference | 0.1 | 스플라이싱 변화 크기 기준 | |
| Option | String | event_subtype | RI,A5SS,A3SS,MXE,SE | 스플라이싱 이벤트 하위 유형 RI : Retained Intron A5SS: Alternative 5’ Splice Site A3SS: Alternative 3’ Splice Site MXE: Mutually Exclusive Exon SE: Skipped Exon |
결과
-
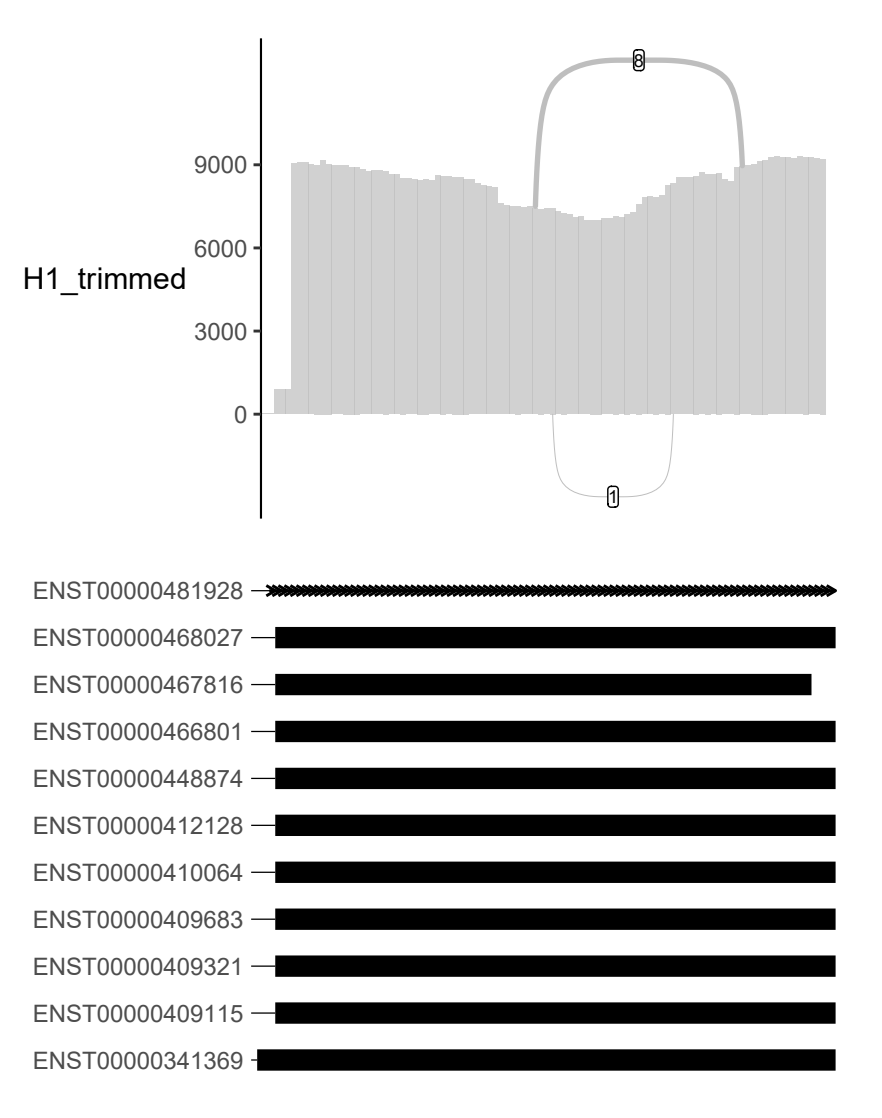
지정한 영역 대한 각각의 ggsashimi plot이 pdf 파일로 생성됨. 첫 번째 plot은 H1 샘플의 coverage plot임. read의 발현 수준을 막대 그래프로 나타냈고, 인트론 스플라이싱 위치를 선으로 시각화함. 2번째 plot은 Trasncript Isoform Tracks으로, 타겟 영역의 기존에 알려진 Trasncript Isoform 주석 정보를 제공함. ENST는 Ensembl에서 사용하는 Transcript ID임.