- 카테고리 Genomics > Variant-analysis
- 수정일2025-10-30 10:48:23
- 레퍼런스
Bio-Express Germline WGS Pipeline은 전장 유전체 시퀀싱 데이터로부터 생식세포 변이를 검출하기 위한 모듈식 분석 파이프라인입니다. 이 파이프라인은 raw FASTQ 파일을 입력으로 사용하고, 개체 유전체 분석을 기반으로 허눈 포괄적인 생식세포 변이 호출 결과와 품질 평가 및 시각화를 제공합니다. FastQC를 통한 시퀀싱 품질 평가 후, Cutadapt로 어댑터 제거 및 품질 트리밍을 수행하고, BWA-MEM2 정렬 도구를 사용하여 참조 유전체 서열에 매핑하여 BAM 형식의 정렬 파일을 생성합니다. 이후 GATK 파이프라인을 통해 중복 제거, 매핑 품질 평가, 그리고 저품질 read 필터링을 수행하며 모든 페어 정보가 일치하는지 확인합니다. SAMtools를 활용한 좌표 기준 정렬과 GATK MarkDuplicates를 통한 PCR 중복 제거를 거쳐, GATK BaseRecalibrator와 ApplyBQSR을 사용하여 알려진 변이 사이트 정보를 공변량으로 활용한 염기 품질 점수 재보정을 수행합니다. 재보정이 완료된 BAM 파일에 대해 먼저 포괄적인 품질 관리 및 샘플 검증 단계를 수행합니다. Somalier를 통한 샘플 관계 검증, VerifyBamID2를 통한 샘플 오염도 평가, 그리고 Mosdepth를 사용한 커버리지 분석을 통해 시퀀싱 데이터의 품질과 신뢰성을 종합적으로 평가합니다. 이어서 GATK HaplotypeCaller를 이용한 GVCF 파일 생성 및 GenotypeGVCFs를 활용한 표준 VCF 형태의 생식세포 SNV/Indel 변이 탐지를 실행합니다. 후속적으로 BCFtools를 적용한 종합적인 변이 통계 해석을 진행하며, Manta 도구를 통해 구조적 변이를 검출합니다.
> 기본 참조 게놈: hg38
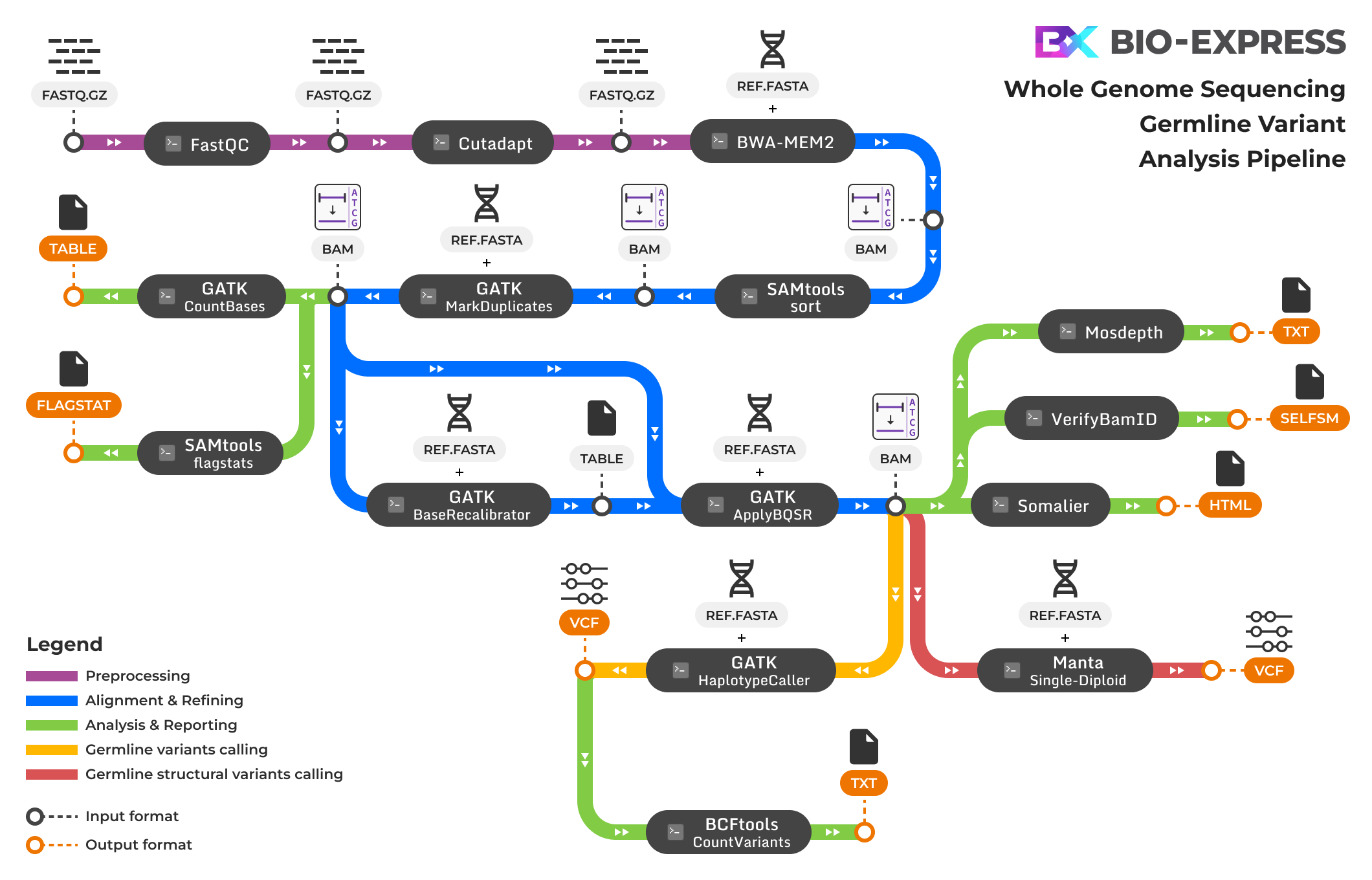
파이프라인 모듈
FastQC
FastQC는 고속 염기서열 분석(high throughput sequence) 데이터의 품질 관리를 위한 분석도구입니다. 이 프로그램은 FASTQ 형식의 서열 데이터를 읽어들여 여러 품질 관리(Qaulity Control) 검사를 수행하고 결과는 HTML 기반의 보고서로 출력합니다. FastQC는 전반적인 품질 문제에 대한 개요 정보를 제공하며, 쉽게 확인할 수 있는 요약된 그래프와 테이블을 포함합니다. FastQC는 FASTQ 형식의 파일이 입력 파일로 사용되며, 출력 결과는 리포트 html 파일과 zip 형식의 압축 파일이 생성됩니다.
주요사항
- FastQC는 자바 애플리케이션입니다. 실행하기 위해서는 시스템에 적절한 자바 실행 환경(Java Runtime Environment, JRE)이 설치되어 있어야 합니다. 따라서 FastQC를 실행하기 전에 먼저 적절한 JRE가 설치되어 있는지 확인해야 합니다. 다양한 종류의 JRE를 사용할 수 있지만, 저희가 테스트해본 것은 최신 오라클 런타임 환경과 adoptOpenJDK 프로젝트의 JRE입니다. 64비트 JRE를 다운로드하여 설치하고, 자바 애플리케이션이 시스템 경로(path)에 포함되도록 설정해야 합니다(대부분의 설치 프로그램이 이를 자동으로 처리해줍니다).
실행 명령어 예시
$program_dir/fastqc –t 6 –o $OUTPUT_DIR $INPUT_DIR/$READ
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o) |
결과
-
.png)
Basic Statistics 테이블은 주어진 FASTQ 파일에 대한 간단한 통계적 정보를 제공합니다. 일반적으로 다음과 같은 정보를 포함합니다. Filename : 분석된 파일의 이름 또는 경로 File type : FASTQ 파일의 종류 Encoding : 품질 점수 인코딩 방식 Total Sequence : 총 서열 수 Filtered Sequences : Read 품질이 좋지 않은 서열 수 Sequence length : 서열의 길이 %GC : 서열에서의 GC 백분율
-
.png)
X축은 리드의 염기 위치를 나타내며, Y축은 품질 점수를 의미합니다. 점수가 높을수록 품질이 좋습니다. 중앙의 빨간색 선은 중앙값을 나타내고, 노란색 박스는 사분위간 범위(25~75%)를 의미합니다. 위쪽 및 아래쪽의 위스커(whisker)는 각각 10% 및 90% 포인트를 나타냅니다. 파란색 선은 평균 품질을 의미합니다. 어떤 염기의 하위 사분위수가 10 미만이거나 중위값이 25보다 작을 경우 경고로 간주됩니다. 또한, 어떤 염기의 하위 사분위수가 5 미만이거나 중위값이 20보다 작으면 오류로 간주됩니다.
-
.png)
색을 이용하여 각 타일의 품질을 나타내며, 파란색은 품질이 높음을 나타내고 빨간색은 품질이 낮음을 나타냅니다. 각 타일의 품질을 모든 타일의 평균 품질과 비교하여 예상 패턴과의 편차를 식별 할 수 있습니다. 특정 타일의 품질이 지속적으로 좋지 않으면 물리적 결함이나 오염 등 셀의 특정 영역에 문제가 있음을 나타낼 수 있습니다. 이상적으로는 모든 타일이 높은 품질을 보여야 하면 플롯에서 더 차가운 색상으로 표시됩니다. 이 플롯은 Illumina 라이브러리에서만 나타납니다.
-
.png)
X축은 리드의 전체 길이에 대한 평균 품질 점수를 나타내고, Y축은 해당 품질 점수를 갖는 읽기의 수를 나타냅니다. 시퀀스의 시퀀싱 품질은 해당 시퀀스에 대한 정확성과 신뢰성을 나타내며, 품질 점수가 높을수록 오류 발생 가능성이 낮다는 것을 의미합니다. 만약 시퀀싱 실행이 전반적으로 낮은 품질을 보인다며, 시퀀싱 화학 문제나 샘플 준비 문제 등이 있을 수 있습니다. 품질 점수가 기록되지 않은 BAM/SAM 파일의 경우 확인할 수 없습니다. 품질 점수가 Phred 척도 기준 최고 품질 점수 27점(오류율 0.2%) 미만일 경우 경고 발생, 20점(오류율 1%) 미만일 경우 오류입니다.
-
.png)
X축은 리드의 포지션을 나타내고, Y축은 시퀀싱한 리드에서 각 base의 전체 비율을 나타냅니다. 좋은 품질의 시퀀싱 샘플에서는 각 위치의 염기 비율을 나타내는 4개의 선이 평행하고 서로 가까워야 합니다. 그러나 선이 일부 위치에서 엉키거나 얽히면 과도하게 표현된 시퀀스가 오염되었음을 나타낼 수 있습니다. 또한, A/T 또는 G/C 염기의 비율이 어떤 위치에서는 10% 이상 차이나면 경고 발생, 20%를 초과하면 오류입니다.
-
.png)
시퀀스에서 G와 C 뉴클레오티드의 백분율 비율을 나타냅니다. 이를 통해 DNA 또는 RNA 시퀀스의 특성을 이해할 수 있습니다. 시퀀스의 GC 함량은 DNA 안정성, 서열의 물리적 특성, 유전자 발현에 영향을 미칠 수 있으므로 중요한 지표 중 하나입니다. X축은 GC contents의 비율을 나타내고, Y축은 시퀀스의 총량을 나타냅니다. 정규 분포와 편차 합계가 전체 리드의 15%를 초과하면 경고, 30%를 초과하면 오류입니다.
-
.png)
시퀀싱 리드의 각 위치에서 발견된 N 비율을 나타내며 일반적으로 매우 낮습니다. 그러나 어떤 위치에서는 N 비율이 5%를 초과하면 시퀀싱 시스템에 문제가 있을 수 있다는 경고로 간주되며, 20%를 초과하면 오류로 간주됩니다. 데이터의 품질이 높은지 확인하고 시퀀싱 읽기의 정확성에 영향을 줄 수 있는 문제를 식별하려면 시퀀싱 중에 N 비율을 모니터링하는 것이 중요합니다. N 비율이 권장 임계값을 초과하는 경우 문제의 심층적인 분석이 필요할 수 있습니다.
-
.png)
시퀀싱 데이터에서 각 리드의 길이에 대한 분포를 나타냅니다. 이 그래프의 X 축은 시퀀스 길이를, Y 축은 리드 수를 나타냅니다. 시퀀싱 데이터에서 발견된 리드의 길이가 어떻게 분포되어 있는지를 시각적으로 확인할 수 있습니다. 이 그래프를 통해 시퀀싱 데이터 세트의 리드 길이 분포를 파악할 수 있으며, 예상치 못한 리드 길이나 이상한 분포를 감지하여 데이터의 품질을 평가하는 데 도움이 됩니다. 일반적으로 시퀀스 길이 분포는 일정하거나 특정한 패턴을 따르지만, 비정상적인 분포는 시퀀싱 데이터에 문제가 있을 수 있다는 신호일 수 있습니다.
-
.png)
중복된 시퀀스가 전체의 20% 이상일 경우 경고, 50% 이상일 경우 오류입니다.
-
.png)
시퀀싱 데이터에서 빈번하게 등장하는 시퀀스를 나타내는 테이블입니다. 이 테이블은 시퀀싱 데이터에서 특정 시퀀스가 기대보다 더 자주 나타나는 경우를 식별합니다. 실험과정에서 발생한 오류, PCR 이중성, 어댑터 오류 또는 샘플의 비정상적인 특정으로 인해 발생할 수 있습니다. 해당 내용을 확인하고 잠재적인 문제를 식별하는 것은 시퀀싱 데이터의 정확성과 신뢰성을 높이는 데 도움이 됩니다.
-
.png)
시퀀싱 데이터에서 발견된 어댑터 시퀀스의 누적 백분율을 보여주는 차트입니다. 시퀀스가 발견된 위치에 따라 백분율이 증가하며, 시퀀스가 리드의 끝까지 존재하는 동안 계산됩니다. 어댑터의 비율을 확인하여 데이터의 품질을 평가하며, 어댑터 시퀀스가 발견되는 비율이 높을수록 시퀀싱 데이터에 오류가 포함될 가능성이 높아지므로, 5%를 초과하면 경고, 10%를 초과하면 오류입니다.
Cutadapt
Cutadapt는 NGS 시퀀싱 데이터에서 어댑터(adapter) 서열 제거, 품질이 낮은 서열 제거 등 다양한 전처리 작업을 수행하는 데 사용되는 도구입니다. 주로 서열의 3’ 말단에서 염기서열을 제거하며, 특히 RNA 시퀀싱 또는 DNA 시퀀싱 데이터에서 어댑터 제거에 빈번하게 활용됩니다. 전체 데이터의 품질을 유지하여 이후의 분석 단계에서 정확한 결과를 얻을 수 있도록 도움을 줍니다. 입력 데이터로는 fastq, fastq.gz, fq, fq.gz 확장자를 가진 파일을 사용할 수 있습니다. 출력으로는 파일 확장자 앞쪽에 ‘_trimmed’라는 문구가 붙은 fastq 파일이 생성되도록 세팅되어 있습니다. 따라서 "trimmed.fastq" 파일은 프라이머 잔여물이나 다른 불순물이 제거된 reads 파일을 나타냅니다.
주요사항
- 1. Bio-Express의 Cutadapt 모듈은 paired-end와 single-end 형식의 FASTQ 데이터를 모두 처리할 수 있도록 설계되었습니다.
- 2. -a 및 –A 옵션에 입력해야 할 각각의 어댑터 서열은 NGS 시퀀서 플랫폼별로 다음과 같습니다. > Illumina: 기본값 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT > MGI: 필요시 기본값 대체 입력 -a AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA -A AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG
실행 명령어 예시
$PROGRAM_DIR/cutadapt -q 20 –Q 20 -j 6 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 --pair-filter=$PAIR_FILTER -o $OUTPUT_DIR/$READ_1 –p $OUTPUT_DIR/$READ_2 $INPUT_DIR/$READ_1 $INPUT_DIR/$READ_2
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o, -p) | |
| Option | Integer | min_len_r1 | 70 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read1의 최소 길이 | |
| Option | Integer | min_len_r2 | 1 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read2의 최소 길이 | |
| Option | String | pair_filter | any | --pair-filter (single-end FASTQ에는 미적용) - any: 두 Read 중 어느 하나라도 조건에 부합하면 필터링 - both: 두 Read 모두 조건에 부합하면 필터링 | |
| Option | String | adapter_pos | adapter | 어댑터 시퀀스 처리 위치 지정 --adapter: 서열의 3' 끝 방향에서 어댑터를 찾아 해당 어댑터와 그 이후 모든 서열을 제거 (-a, -A) --front: 서열의 5' 시작 방향에서 어댑터를 찾아 해당 어댑터와 그 이전 모든 서열을 제거 (-g, -G) --anywhere: 5' 또는 3' 어느 쪽에든 나타날 수 있는 어댑터를 탐지 (-b, -B) | |
| Option | String | adapter_r1 | AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | 첫 번째 Read의 어댑터 시퀀스 (-a) | |
| Option | String | adapter_r2 | AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT | 두 번째 Read의 어댑터 시퀀스 (-A) | |
| Option | Integer | quality_r1 | 20 | 첫 번째 Read의 절단 기준으로 사용할 품질 임계값 (-q) | |
| Option | Integer | quality_r2 | 20 | 두 번째 Read의 절단 기준으로 사용할 품질 임계값 (-Q) |
결과
-
.png)
총 입력된 read pair 수, 어댑터가 검출된 read의 수와 비율, 필터링 후 최종적으로 출력된 read 수 등 전체 리드 처리 현황을 요약함. 또한 데이터 전처리 단계에서 몇 %의 리드가 어댑터에 의해 잘렸고, 몇 %가 최종 분석에 사용 가능한지, 데이터 손실이 얼마나 발생했는지 파악함.
-
.png)
어댑터 서열 (Illumina adapter 등), 탐지 및 제거된 횟수, 최소 overlap 길이, mismatch 허용 개수 (error tolerance), 제거된 어댑터 서열에서의 염기 비율(어댑터 클리핑의 정확성 평가에 활용) 등 Read1에서 탐지된 어댑터의 정보. Read2에 대한 내용도 동일한 방식으로 출력함.
BWA-MEM
BWA는 대용량 시퀀싱 데이터를 참조 유전체와 정렬하는 데 사용되는 고속, 고정밀 분석 도구입니다. BWA는 짧은 리드(30bp~1Mbp)와 긴 리드(>100bp)에 모두 적합하며, 주로 Illumina 시퀀싱 데이터를 정렬하는 데 사용됩니다. BWA의 주요 기능은 효율적인 시퀀스 정렬도, Burrows-Wheeler 변환과 FM 인덱스를 사용하여 시퀀싱 리드를 참조 유전체에 빠르고 정확하게 정렬합니다. BWA-bactrack, BWA-sw 및 BWA-mem의 세 가지 알고리즘으로 구성됩니다. BWA-mem은 긴 리드(>70bp)에 최적화되어 있습니다. 시드-확장 접근 방식을 사용하여 정확하고 효율적인 정렬을 수행하며, 특히 긴 리드에서 높은 성능을 보입니다. 또한 BWA-mem은 RNS-seq 데이터에서 인트론과 엑손 간의 정확한 정렬을 지원하며, 높은 민감도와 정확도를 가지고 있어 변이 탐지에 유리합니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 BWA-mem의 입력(input) 데이터로는 fastq 타입의 파일이 가능하며, 출력 데이터로는 align이 완료된 sam 파일이 생성됩니다.
주요사항
- 1. '-R <문자열>'에서는 완전한 Read Group 헤더 라인을 지정해야 합니다. 이는 후속 분석에서 샘플을 구별하는 데 매우 중요하며, Bio-Express 모듈에서는 아래와 같은 규칙으로 자동 입력됩니다. -R "@RG\\tID:${clean_name}\\tSM:${clean_name}\\tPL:ILLUMINA\\tLB:UNKNOWN" ${clean_name}: '_trimmed', '_R1', '_R2' 등과 같은 불필요한 접미사들은 제거된 샘플명 2. Reference FASTA 파일의 BWA-MEM 프로그램 전용 index가 존재하지 않는다면, reference FASTA 파일로부터 해당 index를 자동으로 생성하여 진행시키도록 설계되어 있습니다. 3. 입력 파일로 사용되는 FASTQ 파일명에 '_trimmed' 또는 '_HQ'와 같은 문구가 있을 경우, 결과 파일명에서 해당 문구는 제거됩니다.
실행 명령어 예시
#BWA-MEM $PROGRAM_DIR/bwa mem –M -v 3 –t 6 –K 100000000 –R "@RG\\tID:${clean_name}\\tSM:${clean_name}\\tPL:ILLUMINA\tLB:UNKNOWN" $REFERENCE_GENOME $READ_1 $READ_2 > $OUTPUT_SAM
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | /path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | /path/to/output_dir | Reference genome FASTA에 FASTQ 데이터가 매핑되어 생성된 SAM/BAM 파일이 생성될 경로 | |
| Option | File | ref_genome | hg38 | Reference genome FASTA 파일. BWA index의 basename. |
결과
-
.png)
SAM 파일은 Sequence alignment data를 담고 있는 텍스트 파일(.txt)로 각 내용은 탭(tab)으로 분리되어 alignment, mapping 정보를 담고 있습니다. Next-generation sequencing (NGS) 을 통해 시퀀싱 된 서열의 전사체 혹은 유전체 서열(Reference, @로 시작되는)에 FASTQ 파일을 다시 mapping 시킨 형태의 파일입니다. SAM 파일의 Header 부분에는 각 Chromosome에 mapping 된 정보를 가지고 있으며, Align 된 read 들은 각 alignment 당 한 줄로 보입니다. @HEADER: Reference로 이용된 서열의 정보, @표시로 시작 @SQ: Header 레코드 시작을 나타냄 SN: 시퀸스 이름을 나타냄 LN: 시퀸스 길이를 나타냄 HWI-EAS038:6:1:23:122#0: 리드의 고유 식별자를 나타냄 4: 리드의 상태를 의미. 여기서 ‘4는’ 리드가 매핑되지 않았음을 의미 TAGCCTTGATGTTTACC..: 리드의 염기서열을 의미 OJYMXLTPKOPOXYBBB...: 리드의 품질 값. Quality scores를 나타냄 HWI-EAS038:6:1:25:283#0: 다른 리드의 고유 식별자 0: 리드의 상태로, ‘0’은 리드가 매핑되었음을 의미 chr14: 리드가 매핑된 참조 서열의 이름 27002726: 리드가 매핑된 위치를 의미 33M: 리드가 33bp만큼 매핑되었음을 의미 AGAGACCCAGGAAATTG..: 리드의 염기 서열을 의미 abaaa_Z_X]PM*BBBBBB..: 리드의 품질값을 의미
SAMtools_sort
SAMtools는 고처리량 시퀀싱 데이터를 조작하고 분석하기 위해 설계된 다목적 도구 모음으로, 특히 SAM, BAM, CRAM 형식의 정렬 데이터를 처리하는 데 사용됩니다. 이 도구는 변이 호출, 정렬 데이터 확인, 품질 관리 등 다양한 작업에 널리 활용됩니다. SAMtools는 인덱싱, 정렬, 병합, 필터링 등 정렬 파일 처리에 필요한 다양한 기능을 지원하며, 유전체 분석 파이프라인에서 필수적인 도구입니다.SAMtools sort 명령은 SAM, BAM, CRAM 파일을 지정된 기준에 따라 정렬합니다. 기본적으로 좌표(coordinate) 기준으로 정렬하며, -n 옵션을 사용하면 쿼리 이름(query name) 기준으로 정렬합니다. 정렬 순서는 SAM 파일 헤더의 @HD 태그 내 SO 필드에 표시됩니다. 좌표 정렬은 @HD SO:coordinate, 쿼리 이름 정렬은 @HD SO:queryname으로 헤더에 기록됩니다.
좌표 정렬 (Coordinate Sorting)의 경우, 읽기는 다음과 같은 순서로 정렬됩니다:
1. 참조 시퀀스 이름(RNAME): @SQ 태그에 정의된 참조 시퀀스 사전의 순서를 따릅니다.
2. 가장 왼쪽 매핑 위치(POS): 동일한 RNAME 내에서 읽기의 시작 위치를 기준으로 정렬됩니다.
3. REVERSE 플래그: POS가 동일한 경우, 순방향 가닥(forward strand, REVERSE 플래그 0)이 역방향 가닥(reverse strand, REVERSE 플래그 1)보다 먼저 옵니다.
이후 추가적인 동점이 있는 경우, 정렬 순서는 입력 데이터의 순서를 유지할 수 있습니다.
쿼리 이름 정렬 (Query Name Sorting)의 경우, -n 옵션을 사용하며 읽기는 다음과 같은 순서로 정렬됩니다:
1. 쿼리 이름(QNAME): 자연스러운 순서(natural order)로 정렬되며, 문자열 내 숫자 부분은 수치적으로 비교됩니다 (예: "read9"는 "read10"보다 먼저).
2. READ1/READ2 플래그: 동일한 QNAME을 가진 읽기는 첫 번째 읽기(READ1, 플래그 0x40)가 두 번째 읽기(READ2, 플래그 0x80)보다 먼저 옵니다.
3. 정렬 유형: READ1/READ2가 동일한 경우, 주 정렬(primary alignment)이 먼저 오고, 그 다음 보조 정렬(supplementary alignment), 2차 정렬(secondary alignment) 순으로 정렬됩니다.
남은 동점은 입력 데이터의 원래 순서를 따릅니다. 따라서 SAMtools는 유전체 데이터를 효율적으로 처리하고 분석하는 데 중요한 역할을 하며, 특히 정렬 작업에서 sort 도구가 유용하게 사용됩니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 SAMtools sort는 SAM파일을 입력 데이터로 사용하여 BAM 파일을 출력합니다.
실행 명령어 예시
$PROGRAM_DIR/samtools sort -@ 6 –O bam —write-index –o $OUTPUT_BAM $INPUT_BAM
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | /path/to/input_dir | Sorting이 필요한 BAM 파일을 찾을 디렉토리 | |
| Output | Folder | output_dir | /path/to/output_dir | Sorting이 완료된 BAM 파일을 저장할 디렉토리 |
결과
-

좌표 기준 정렬 전 상태. 모든 레코드의 RNAME(3열)은 chr1이며, POS(4열) 순서가 10000 → 10002 → 10001 → 10003 처럼 뒤섞여 있음. 둘째~셋째 줄의 chr2 표기는 RNEXT(7열)로, 짝 리드의 위치 정보일 뿐 현재 레코드의 정렬 참조는 아님.
-

좌표 기준 정렬된 상태. 모든 레코드의 RNAME(3열)이 chr1이며, POS(4열)이 10000 → 10001 → 10002 → 10003 순으로 오름차순. 둘째 줄의 chr2는 RNEXT(7열)로, paired 리드가 chr2에 매핑됨을 의미(해당 레코드는 여전히 chr1에 정렬). RNEXT가 =인 경우는 paired 리드가 같은 염색체(여기서는 chr1)에 정렬되어 있다는 뜻.
GATK MarkDuplicates
GATK의 MarkDuplicates는 BAM 또는 SAM 파일에서 중복 리드를 식별하고 태그하는 분석 도구입니다. BAM/SAM 파일에서 리드와 리드 페어의 5' 위치의 시퀀스를 비교하여 중복을 식별합니다. 중복된 리드를 모은 후에는 리드의 베이스 품질 점수의 합을 기준으로 주 리드와 중복 리드를 구별하는 알고리즘을 사용하여 표시합니다. 이러한 중복 서열은 변이 검출 시 위양성(false positive)을 유발할 수 있으므로, 제거하거나 통계 분석에서 제외하는 것이 중요합니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 GATK의 MarkDuplicates의 입력(input) 데이터로는 BAM 파일이며, 중복이 식별된 리드가 표시된 새로운 BAM 파일과 중복된 리드의 수와 관련된 다양한 통계를 담은 메트릭스 파일을 출력(output) 데이터로 합니다.
실행 명령어 예시
$PROGRAM_DIR/gatk MarkDuplicates \ -I $INPUT_BAM -O $OUPUT_BAM -R $REFERENCE_GENOME -M $METRICS_file
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | 중복 서열 제거 처리 대상 BAM 파일을 찾을 디렉토리 | |
| Output | Folder | output_dir | path/to/output_dir | Sorting이 완료된 BAM 파일을 저장할 디렉토리 | |
| Option | File | ref_genome | hg38 | Reference genome FASTA 파일 |
결과
-
.png)
## METRICS CLASS LIBRARY: 분석된 라이브러리 UNPAIRED_READS_EXAMINED: 분석된 단독 리드의 총 수 READ_PAIRS_EXAMINED: 분석된 리드 페어의 총 수 SECONDARY_OR_SUPPLEMENTARY_RDS: 보조 또는 부가 정렬된 리드의 수 UNMAPPED_READS: 매핑되지 않은 리드의 수 UNPAIRED_READ_DUPLICATES: 단독 리드의 중복 수 READ_PAIR_DUPLICATES: 중복된 리드 페어의 수 READ_PAIR_OPTICAL_DUPLICATES: 시퀀싱 기기의 광학 센서가 잘못 인식하여 발생한 중복 리드 페어의 수 PERCENT_DUPLICATION: 전체 리드 중 중복된 리드의 백분율 ESTIMATED_LIBRARY_SIZE: 중복 리드의 수를 바탕으로 계산된 시퀀싱 라이브러리 규모 ## HISTOGRAM BIN: 리드 페어의 총 중복 수를 구간별로 나눔 CoverageMult: 리드 커버리지의 배수, 실험의 리드 깊이를 평가하는데 사용
GATK CountBases
GATK의 CountBases는 BAM 또는 SAM 파일에서 각 염기(A, C, G, T)와 기타 염기 외 문자(N 등)의 출현 빈도를 계수하는 분석 도구입니다. 시퀀싱 데이터의 품질을 평가하거나 유전자 발현 수준, 변이 분석 등 다양한 분석 과정에서 유용하게 사용됩니다. CountBases의 출력은 초기 데이터 탐색 단계에서 데이터의 일관성과 정확성을 평가하는 데 중요한 지표를 제공하며, 이를 기반으로 추가적인 실험 설계나 분석 전략을 결정할 수 있습니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 GATK의 CountBases는 입력(input) 데이터로 BAM 파일을, 출력(output) 데이터로는 BAM 파일 전체 bases 수가 출력된 count 파일이 생성됩니다.
실행 명령어 예시
$program_dir/gatk CountBases -I $INPUT_BAM -O $OUTPUT_DIR -R $REFERENCE_GENOME -L $GENOMIC_INTERVALS -RF NotSecondaryAlignmentReadFilter
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | 염기 개수를 계산할 BAM 파일을 찾을 디렉토리 | |
| Output | Folder | output_dir | path/to/output_dir | 염기 개수 계산 결과물을 저장할 디렉토리 | |
| Option | File | ref_genome | hg38 | Reference genome FASTA 파일 (-R) | |
| Option | File | genomic_intervals | hg38 | Genomic Interval 파일: 본 분석 과정을 수행할 특정 유전체 영역을 지정 (-L) |
결과
-

총 염기 수가 161,513,787,863 라는 것은, human WGS 데이터일 경우, 평균적으로 각 염기 위치가 약 54회 시퀀싱되었다는 의미임. > 평균 커버리지: 161,513,787,863 (총 염기 수) / 약 30억개 base (인간 게놈 크기) ≈ 54X
SAMtools_flagstat
SAMtools flagstat는 BAM, SAM, CRAM 파일의 FLAG 정보를 기반으로 정렬 통계를 계산하고 요약 보고서를 제공하는 품질 관리 도구입니다. 이 도구는 입력 파일을 한 번 완전히 통과하면서 FLAG 필드의 비트 플래그를 기준으로 13개 카테고리의 통계를 산출하여 표준 출력으로 결과를 제공합니다. FLAG 정보는 SAM 형식 사양서에 정의된 비트 플래그로, 각 리드의 정렬 상태와 특성을 나타내는 중요한 메타데이터입니다. flagstat는 전체 서열 수, 매핑된 서열 수, 쌍으로 매핑된(properly paired) 서열 수, 중복(duplicates) 리드 수, 주 정렬(primary), 보조 정렬(secondary), 보완 정렬(supplementary) 등의 세부 통계를 제공합니다. 각 카테고리는 QC 통과(QC-passed)와 QC 실패(QC-failed) 리드로 구분되어 "#PASS + #FAIL" 형태로 출력되며, 매핑 비율, 적절한 페어링 비율 등의 백분율 정보도 함께 제공됩니다. 또한 기본 형식 외에도 TSV(탭으로 구분된 값)와 JSON 형식으로 출력할 수 있어 스프레드시트 소프트웨어나 다른 분석 도구에서 쉽게 활용할 수 있습니다. 이러한 통계 정보는 시퀀싱 실험의 품질을 평가하고, 정렬 과정의 성공률을 확인하며, 데이터의 전반적인 특성을 파악하는 데 필수적인 지표로 활용됩니다. 특히 매핑률이 낮거나 적절하게 페어링되지 않은 리드의 비율이 높은 경우, 실험 조건이나 분석 파라미터를 재검토할 필요가 있음을 시사합니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 SAMtools flagstat는 입력(input) 데이터로 BAM 파일을 사용하며, 출력(output) 데이터로는 정렬 통계가 포함된 텍스트 형태의 요약 보고서를 생성합니다.
실행 명령어 예시
$PROGRAM_DIR/samtools flagstat -@ 6 \ -I $INPUT_BAM \ > $OUTPUT_FILE
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 |
결과
-

품질 평가 기준 - 매핑률(mapped %): >95% 권장 - Properly paired: >90% 권장 - 중복률(duplicates): <30% 권장 (WGS 기준)
GATK BaseRecalibrator
GATK의 BaseRecalibrator는 리드의 염기 품질 점수와 오류 패턴을 모델링하기 위한 데이터를 수집합니다. 이 단계에서는 BAM 파일과 신뢰할 수 있는 변이 목록을 입력으로 받아, 리드의 품질 점수를 재조정하는 데 필요한 정보를 수집합니다. BaseRecalibrator는 다양한 공변랑을 기반으로 테이블을 생성합니다. 탐색은 알려진 변이 사이트 VCF 파일에 있는 위치에만 작동합니다. 모든 참조 불일치를 오류로 간주하고, 이를 통해 낮은 베이스 품질을 나타낸다고 가정합니다. 충분한 데이터를 통해 특정 공변량이 관찰된 사이트에서 경험적 오류 확률을 계산할 수 있습니다. 오류 확률은 불일치 횟수를 관찰 횟수로 나누어 계산됩니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 GATK의 BaseRecalibrator는 입력(input) 데이터로 BAM 파일, 참조 유전체 서열을 포함하는 FASTA 파일과 신뢰할 수 있는 변이 정보를 포함하는 VCF 파일입니다. 출력(output) 데이터로는 여러 공변량 값, 관찰 횟수, 불일치 횟수 그리고 경험적 품질 점수를 포함하는 table 파일입니다.
주요사항
- > hg38 reference genome 기준, 일반적으로 사용되는 known sites 4개 --known-sites dbsnp_138.hg38.vcf.gz --known-sites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz --known-sites 1000G_phase1.snps.high_confidence.hg38.vcf.gz --known-sites Homo_sapiens_assembly38.known_indels.vcf.gz
실행 명령어 예시
$PROGRAM_DIR/gatk BaseRecalibrator -I $INPUT_BAM -O $OUTPUT_TABLE -R $REFERENCE_GENOME --known-sites $KNOWN_SITES_[1234]
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | File | ref_genome | hg38 | Reference sequence FASTA 파일 경로 | |
| Option | File | known_sites_1~4 | hg38 | known polymorohic sites인 VCF 파일 경로 |
결과
-
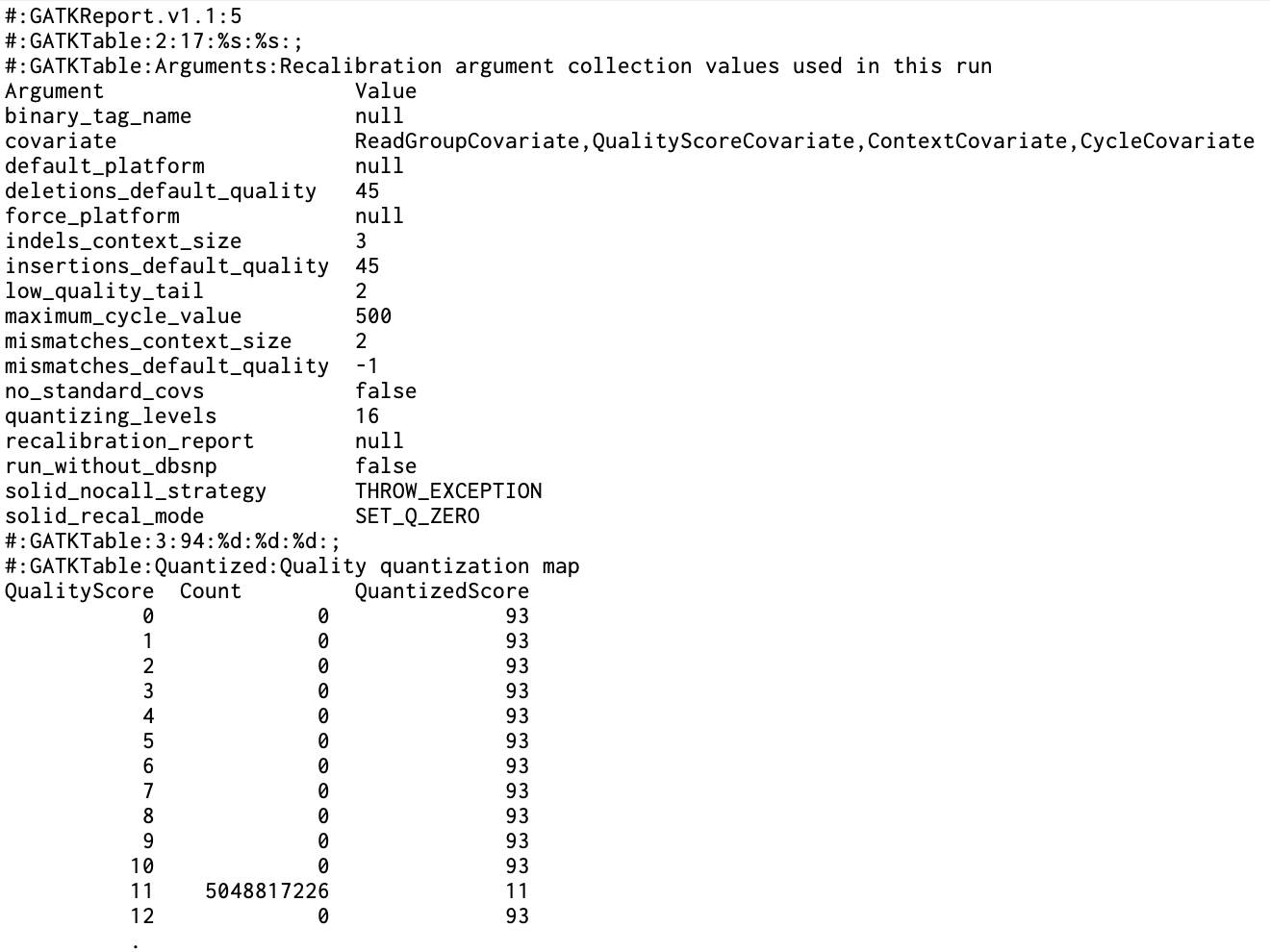
> 파일 헤더 #:GATKReport.v1.1:5 : GATK Report 파일 형식의 버전과 관련된 메타데이터 > Arguments 섹션 Argument: 사용된 인수의 이름 Value: 해당 인수의 값 > Quality quantization map 섹션 QualityScore: 원래 품질 점수 Count: 해당 품질 점수가 나타난 횟수 QuantizedScore: 양자화된 품질 점수
GATK ApplyBQSR
GATK의 ApplyBQSR은 GATK에서 중요한 역할을 하는 도구로, 시퀀싱 데이터의 염기 품질 점수를 재조정하여 변이 탐지의 정확성을 향상시키는 기능을 수행합니다. 이 도구는 “BaseRecalibrator”에서 생성된 재조정 테이블에 포함된 여러 공변량을 사용하여 BAM 파일의 각 염기에 대한 품질 점수를 정밀하게 재조정합니다. 기본 공변량에는 리드 그룹, 초기 품질 점수, 시퀀싱 사이클, 뉴클레오타이드 컨텍스트 등이 포함됩니다. 각 공변량을 통해 특정 염기에서 발생할 수 있는 오류 확률을 계산하고, 이를 바탕으로 새로운 품질 점수를 할당합니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 GATK의 ApplyBQSR은 입력(input) 데이터로 정렬된 BAM 파일, “BaseRecalibrator”를 통해 생성된 재조정 테이블 파일과 참조 유전체 서열을 포함하는 FASTA 파일을 사용합니다. 출력(output) 데이터로는 재조정된 품질 점수를 반영한 새로운 BAM 파일입니다.
실행 명령어 예시
$PROGRAM_DIR/gatk ApplyBQSR -I $INPUT_BAM -bqsr $RECAL_TABLE -O $OUTPUT_BAM -R $REFERENCE_GENOME
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | BAM 파일이 있는 디렉토리 경로 | |
| Input | Folder | table_dir | path/to/table_dir | BaseRecalibrator에 의해 table 파일이 생성된 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | File | ref_genome | hg38 | Reference sequence FASTA 파일 경로 |
결과
-
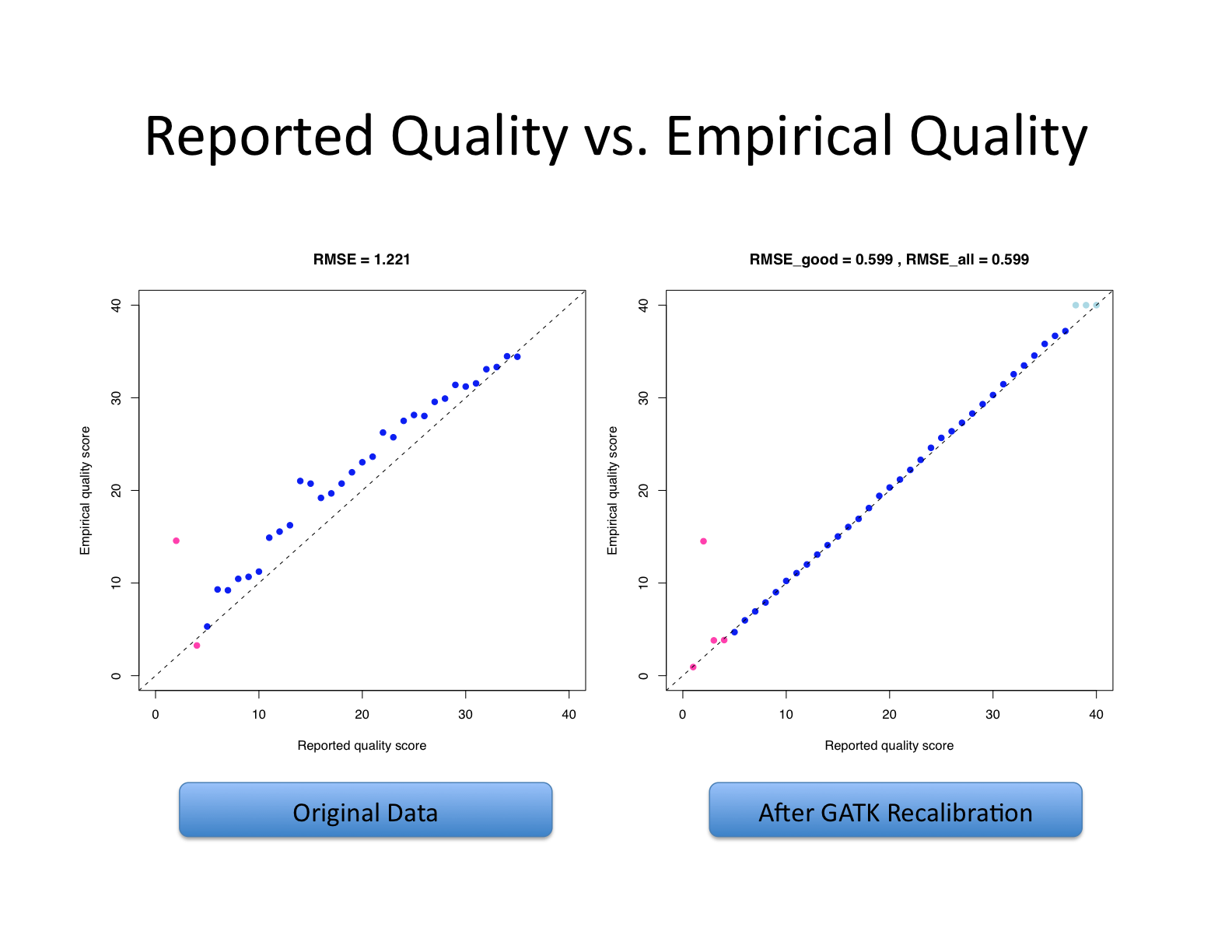
왼쪽 그래프는 원본 데이터의 상태를, 오른쪽 그래프는 GATK 재보정 후의 결과를 보여 나타냄. RMSE_good과 RMSE_all이 모두 0.599로 동일하게 나타나는 것은 전체 데이터에서 일관된 보정 효과가 달성되었음을 의미함. - 출처: https://ucdavis-bioinformatics-training.github.io/2021-July-Genome-Wide-Association-Studies/data_reduction/variantcalling
-
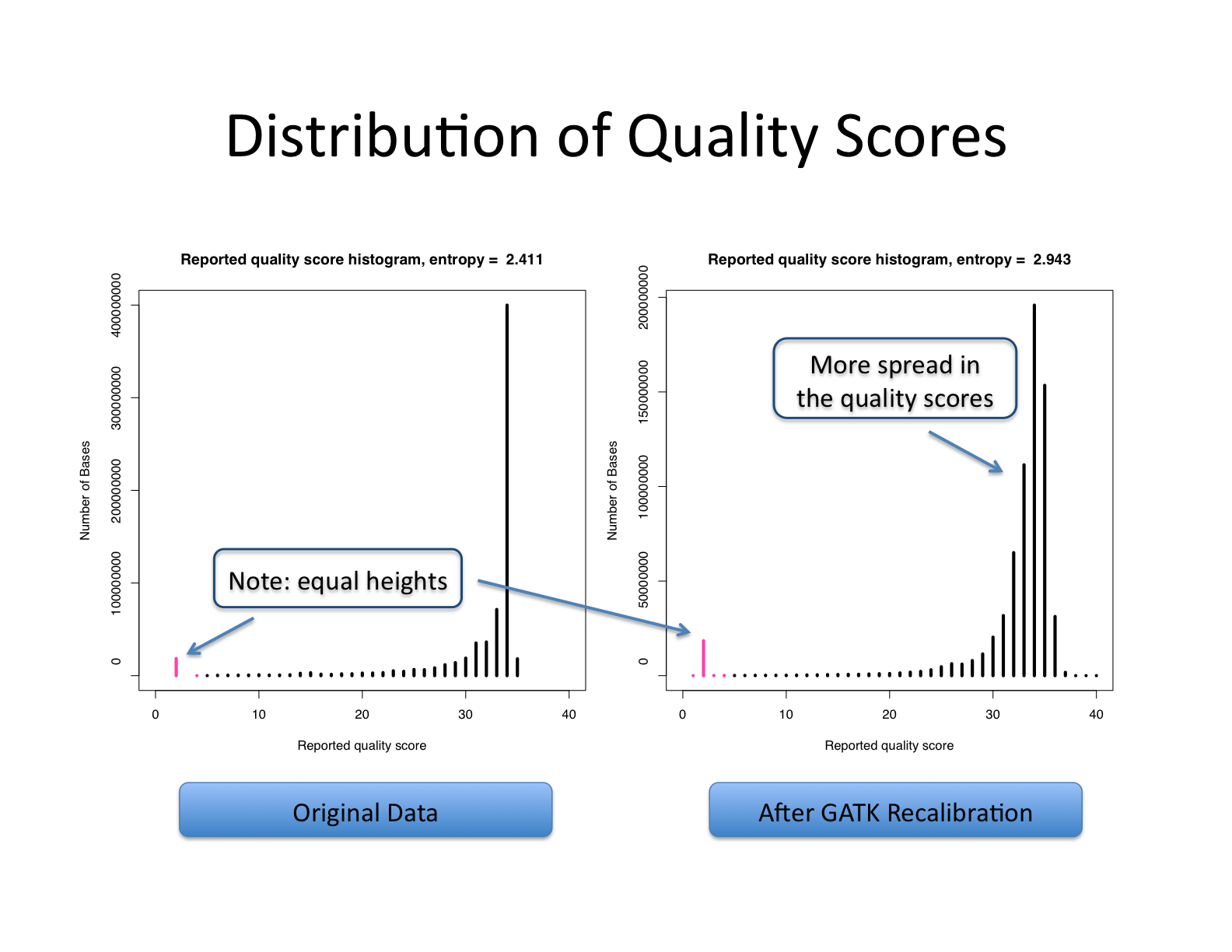
왼쪽 그래프는 원본 데이터(Original Data)의 상태를, 오른쪽 그래프는 GATK 재보정 후의 결과를 보여 나타냄. 원본 데이터에서는 대부분의 염기가 품질 점수 30 근처에 집중되어 있었지만, 재보정 후에는 엔트로피가 2.411에서 2.943으로 증가하여 각 염기의 실제 품질에 따라 더 넓은 범위의 점수로 더 다양하고 정확해졌음. - 출처: https://ucdavis-bioinformatics-training.github.io/2021-July-Genome-Wide-Association-Studies/data_reduction/variantcalling
Somalier
Somalier는 유전체 데이터의 샘플 간 관련성을 분석하고 확인하는 분석 도구입니다. 주로 샘플의 신원을 확인하고 유전체 데이터에서 샘플 간의 혼합이나 오류를 탐지하는 데 사용됩니다. BAM, CRAM, VCF 파일에서 정보 추출을 수행하며, 샘플 간의 유사도 매트릭스를 계산하여 시각화할 수 있습니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 Somalier는 입력(input) 데이터로 BAM 파일, Reference sequence FASTA 파일과 추출할 variant의 sites인 VCF 파일을 사용합니다. 출력(output) 데이터로는 추출된 샘플 정보가 포함된 JSON 형식의 .somalier 파일입니다.
주요사항
- > Relatedness의 의미 - 1.00 : 일란성 쌍둥이/클론/중복 샘플 - 0.50 : 부모-자식/형제자매/부모의 일란성 쌍둥이/일란성 쌍둥이의 자식 - 0.35 : 3/4 형제 (예: 같은 어머니, 아버지들이 형제) - 0.25 : 조부모-손자/이복형제/삼촌-조카/이종사촌 - <0.20 : 원거리 친척(사촌)/혈연 관계 없음
실행 명령어 예시
$program_dir/somalier extract –d $OUTPUT_DIR \ -s $SOMALIER_SITES \ -f $REFERENCE_GENOME \ $INPUT_BAM $program_dir/somalier relate –o $OUTPUT_DIR \ $SOMALIER_EXTRACT_OUTPUT
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | 분석할 BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | File | ref_genome | hg38 | Reference sequence FASTA 파일 경로 | |
| Option | File | somalier_sites | hg38 | 추출할 variant의 sites인 VCF 파일 경로 |
결과
-
.png)
샘플 간 유사도(sample-to-sample relatedness)와 샘플 품질 관련 QC 지표들을 비교/탐색할 수 있게 구성되어, 연구자가 여러 개의 샘플 데이터를 Somalier로 처리했을 때, 각 샘플간 관계성, 유전형의 품질, 성별/성염색체의 ploidy, 유전적 유사성 등을 시각적으로 파악할 수 있도록 하여, 예상치 못한 이상치(outlier), 잘못된 성별 할당, 샘플 처리 오류 등을 조기에 발견할 수 있도록 함. > Sample to Sample Relatedness: shared hets, shared hom-alts, homozygous concordance, relatedness, IBS0, IBS2 등 서로 다른 샘플 간의 유전적 유사도를 나타내는 지표들을 X축과 Y축에 할당함으로써 scatter plot 형태로 비교 > Sample Depth Metrics: allele balance, unknown genotype 비율, 각 유전형(0/0, 0/1, 1/1) 사이트 수, X/Y 염색체 상의 깊이 및 유전형 사이트 수 등 서로 다른 샘플 간의 유전적 유사도를 나타내는 지표들을 X축과 Y축에 할당함으로써 scatter plot 형태로 비교 > Preset selection for sample plot: QC 작업 시 빠르게 유용한 조합의 지표들을 선택 가능하도록, 여러 목적에 맞는 프리셋 옵션을 제공 - Sex QC: X 염색체의 0/1 (이형접합) 사이트 수 vs. chrX의 scaled mean depth - Sex ploidy QC: chrX vs chrY 깊이 비교 - General QC: unknown genotype 비율 vs allele balance가 극단적인 het 비율 - Depth and allele QC: 전체 깊이 vs mean heterozygous allele balance
-

relatedness: 관련성 계수 (관계 정도를 나타내는 수치) IBS0L: 한 샘플은 동형접합 참조형, 다른 샘플은 동형접합 대립형인 사이트 수 IBS2L 두 샘플이 동일한 유전형을 갖는 사이트 수 hets_a: 샘플 a가 이형접합인 사이트 수 hets_b: 샘플 b가 이형접합인 사이트 수 hets_ab: 샘플 a 또는 b가 이형접합인 사이트 수 shared-hets: 두 샘플 모두 이형접합인 사이트 수 shared-hom-alts: 두 샘플 모두 이형접합인 사이트 수 hom_alts_a: 샘플 a가 동형접합 대립형인 사이트 수 hom_alts_b: 샘플 b가 동형접합 대립형인 사이트 수 shared_hom_alts: 두 샘플 모두 동형접합 대립형인 사이트 수 N: 전체 사이트 수 x_ibs0: X 염색체에서의 IBS0 x_ibs2: X 염색체에서의 IBS2
-
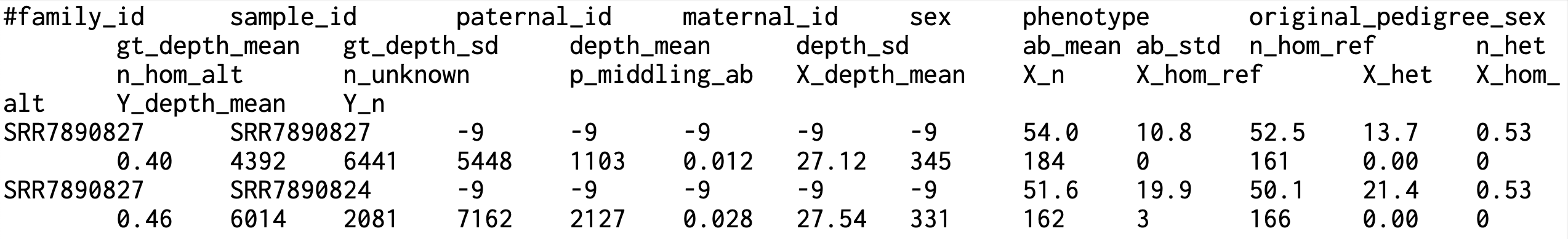
family_id, sample_id, paternal_id, maternal_id, sex, phenotype: 가족/개인/부/모/성별/표현형: PLINK PED 형식과 동일 original_pedigree_sex: 입력한 PED 파일에 적혀 있던 원래의 성별 값(추정/교정 전 기록) gt_depth_mean, gt_depth_sd: Somalier가 유전형을 실제로 판정한 사이트에 대해 계산한 평균 커버리지(깊이)와 표준편차 depth_mean, depth_sd: 검사한 전체 사이트(유전형이 미정인 지점 포함)의 ref+alt 카운트로부터 계산한 평균 깊이와 표준편차 ab_mean, ab_std: 대립유전자 균형(allele balance: AB = alt / (ref+alt)의 평균과 표준편차. 이상적 평균값은 0.5 근처(균형) n_hom_ref, n_het, n_hom_alt, n_unknown: Somalier가 선택한 정보성 변이 좌위에서의 유전형 카운트 - hom-ref: AB < 0.02 - het: 0.2 ≤ AB ≤ 0.8 - hom-alt: AB > 0.98 - unknown: 위 기준에 들지 않음 p_middling_ab: "중간대(middling) AB"에 속하는 지점의 비율. 즉, 이형접합으로 보이는 자리들의 AB가 0.5 근처의 균형대에 머무는 정도를 나타내는 QC 지표. WGS 고품질 샘플에서 높게 나오는 경향 있음. X_depth_mean, X_n, X_hom_ref, X_het, X_hom_alt: X 염색체 상의 정보성 좌위만 따로 모아 계산한 평균 깊이/좌위 수/유전형 카운트. 성별 QC에 활용되며, 일반적으로 XX(여성)는 X에서 이형접합이 많이 관찰되고 X(남성)는 X에서 이형접합이 거의 없음이 기대됨(LOH, 클론성 등 예외 존재) Y_depth_mean, Y_n: Y 염색체 좌위에서의 평균 깊이와 좌위 수. 남성 샘플은 Y-depth가 유의하게 나타나고, 여성은 거의 0에 가까움(정렬/타깃/실험에 따라 예외 가능)
VerifyBamID
VerifyBamID2는 시퀀싱 데이터(BAM/CRAM 파일)에서 DNA 오염도(contamination)를 정확하게 추정하기 위한 차세대 품질 관리 도구입니다. 혈통에 무관한(ancestry-agnostic) 방법을 사용하여 다양한 인종 배경의 샘플에서도 일관되고 신뢰할 수 있는 오염도 측정을 제공합니다. 이 도구는 주성분 분석(Principal Component Analysis, PCA)과 특이값 분해(Singular Value Decomposition, SVD) 기법을 활용하여 의도된 샘플(intended sample)과 오염 소스(contaminating sample)의 유전적 특성을 각각 추정합니다. VerifyBamID2는 베이지안 최적화 알고리즘을 통해 두 소스의 주성분 좌표(PC coordinates)를 동시에 추정하고, 최대우도법(Maximum Likelihood Estimation)을 사용하여 오염 수준(Alpha)을 정량화합니다. VerifyBamID2의 핵심 혁신은 집단 구조(population structure)에 의존하지 않는 분석 방법입니다. 기존 도구들이 특정 인종 집단의 참조 패널에 의존했던 것과 달리, 이 도구는 SVD 기반의 차원 축소를 통해 샘플의 유전적 배경을 자동으로 추론하고 보정합니다. 이를 통해 혼혈 샘플이나 희귀 집단에서도 정확한 오염도 측정이 가능하며, 대규모 다인종 코호트 연구에서 특히 유용합니다. 특히 종양-정상 쌍 분석에서 VerifyBamID2는 정상 샘플에 종양 세포가 혼입되었는지, 또는 종양 샘플에 정상 세포가 과도하게 포함되었는지를 정량적으로 평가할 수 있습니다. 또한 샘플 라벨링 오류, 교차 오염(cross-contamination), 그리고 액체 생검(liquid biopsy)에서의 순환 종양 DNA(ctDNA) 비율 추정에도 활용됩니다.
주요사항
- > SVD_PREFIX 예시: - 1000g.phase3.100k.b38.vcf.gz.dat : hg38 기반 1000 Genomes - 1000g.phase3.100k.b37.vcf.gz.dat : hg19 기반 1000 Genomes - hgdp.100k.b37.vcf.gz.dat : hg19 기반 Human Genome Diversity Project SVD_PREFIX를 1000g.phase3.10k.b38.vcf.gz.dat으로 지정하고자 할 때, 1000g.phase3.10k.b38.vcf.gz.dat.V를 선택하면, “.V”가 자동으로 제거되도록 설계되어 있음.
- > 오염도 평가 기준 - FREEMIX < 0.02 : 정상 범위 - 0.02 ≤ FREEMIX < 0.05 : 경미한 오염 - FREEMIX ≥ 0.05 : 심각한 오염 (분석 주의)
실행 명령어 예시
$PROGRAM_DIR/verifybamid --SVDPrefix $SVD_PREFIX \ --BamFile $INPUT_BAM \ --Reference $REFERENCE_GENOME \ --Output $OUTPUT_PREFIX \ --NumThread 6
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | File | ref_genome | hg38 | Reference sequence FASTA 파일 경로 | |
| Option | File | svd_prefix | 1000g.phase3.10k.b38.vcf.gz.dat.V | Singular Value Decomposition(SVD) 기법으로 준비된, VerifyBamID 전용 참조 패널 파일들(.UD, .mu, .bed, .V)의 prefix |
결과
-

#SNPS: 분석에 사용된 SNP 마커 수 AVG_DP: 평균 시퀀싱 깊이 FREEMIX: 오염도 추정치 (0~1) FREELK1: 오염 가정 하의 우도값 FREELK0: 순수 샘플 가정 하의 우도값
-

Contaminating Sample: 오염 소스의 주성분 좌표 Intended Sample: 의도된 샘플의 주성분 좌표
Mosdepth
Mosdepth는 전장 유전체 시퀀싱(WGS), 엑솜 시퀀싱, 표적 시퀀싱을 위한 고속 BAM/CRAM 커버리지 계산 도구입니다. Nim 언어로 개발되어 기존 도구들보다 월등히 빠른 성능을 제공하며, 메모리 효율적인 알고리즘을 통해 대용량 시퀀싱 데이터의 커버리지 분석을 신속하게 수행합니다. 이 도구는 독특한 염색체별 배열 기반 알고리즘을 사용합니다. 각 염색체에 대해 배열을 생성하고, 리드의 시작 위치에서는 값을 증가시키고 끝 위치에서는 감소시킨 후, 누적합을 계산하여 각 위치의 커버리지를 구합니다. 이 방법은 CIGAR 연산을 정확히 추적하여 리드의 정렬된 모든 부분을 포함하며, 겹치는 mate-pair를 이중 계산하지 않는 정교한 커버리지 측정을 제공합니다. Mosdepth는 단순한 커버리지 계산을 넘어서 다양한 고급 기능을 제공합니다. 윈도우 기반 분석, BED 파일 기반 영역별 분석, 임계값 기반 커버리지 통계, 그리고 quantize 기능을 통한 커버리지 구간 분할 등이 가능합니다. 특히 분포 계산은 런타임 증가 없이 자동으로 수행되며, 다중 스레드를 활용한 압축 해제 최적화로 대규모 데이터셋에서도 뛰어난 성능을 발휘합니다.
주요사항
- > 커버리지 품질 기준 - 평균 깊이 : WGS 30X 이상 권장 - 균일성 : CV(변동계수) < 0.3 - 10X 이상 비율 : >90% - 30X 이상 비율 : >80%
실행 명령어 예시
$PROGRAM_DIR/mosdepth –t 6 $OUTPUT_PREFIX $INPUT_BAM
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 |
결과
-
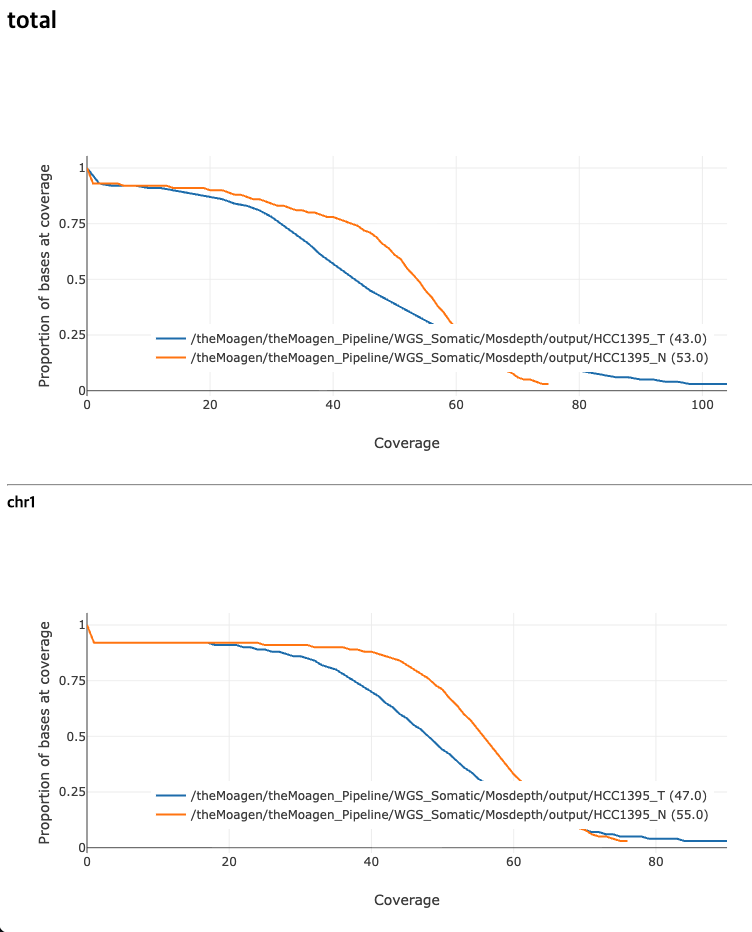
모든 샘플들의 전체 세트 및 염색체별 커버리지 분포 plot
-
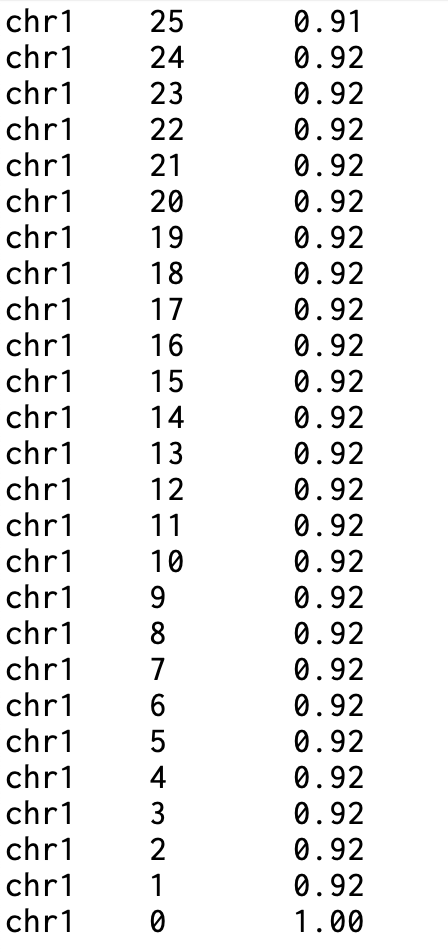
"특정 커버리지 이상으로 덮인 염기의 비율"을, 염색체별 및 전체 합계에 대한 누적 분포로 표현함. - 1. 염색체 이름 및 전체 합계 - 2. 커버리지 값 - 3. 해당 커버리지까지의 비율(0~1) “chr1 30 0.85”: chr1의 염기 중 85%가 ≥30x 이상으로 덮였다는 뜻. 각 염색체 블록의 마지막 줄은 coverage=0에 fraction=1.0(모두 ≥0x)로 끝남.
-
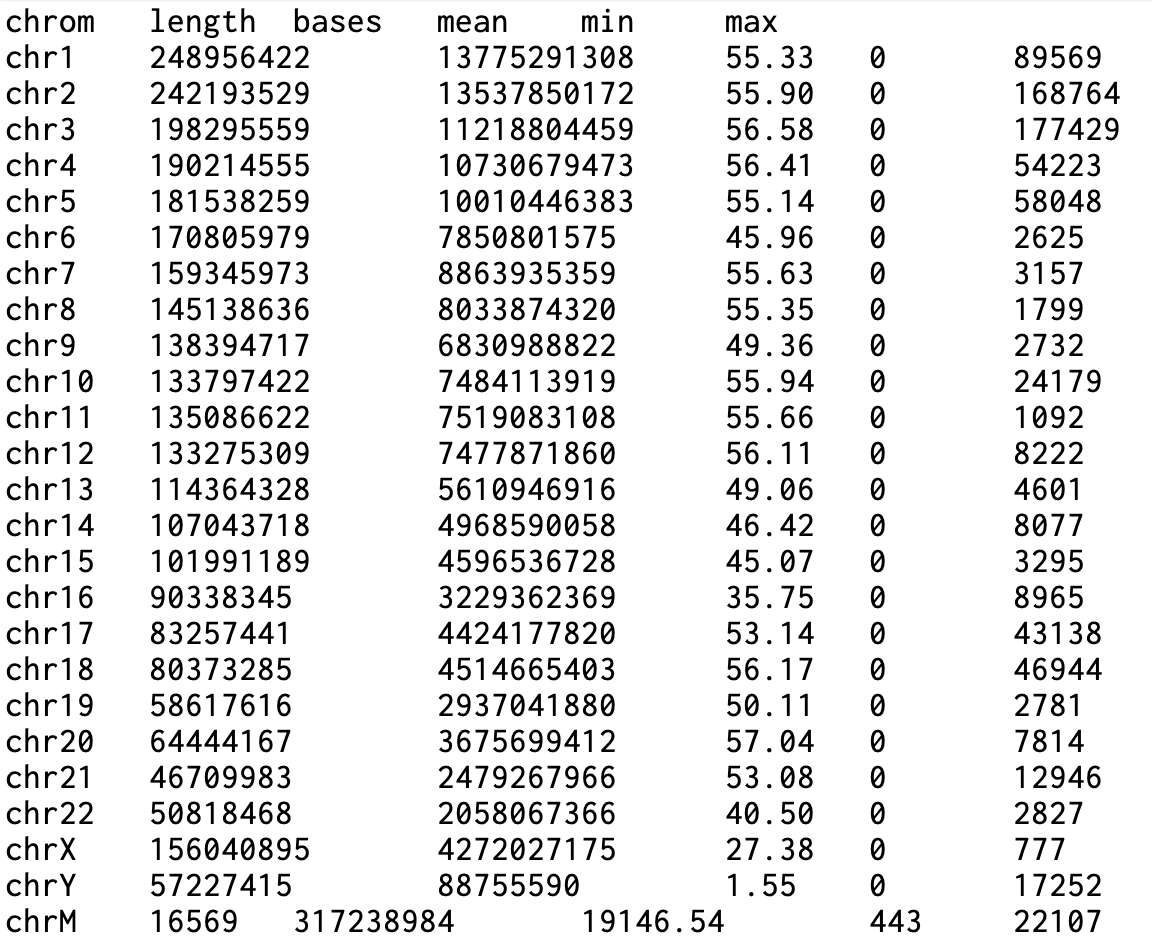
1. chrom: 염색체(또는 total) 2. length: 해당 염색체의 길이(bp) 3. bases: 계산에 포함된 염기 수 4. mean: 평균 커버리지 5. min: 최소 커버리지(관측된 최소값) 6. max: 최대 커버리지(관측된 최대값)
-
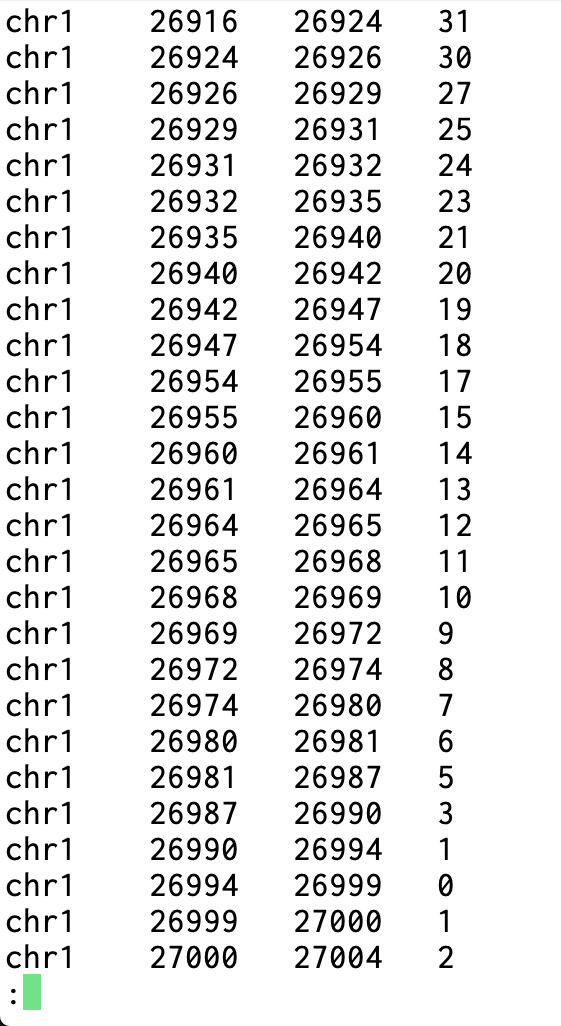
모든 염색체들의 위치별 커버리지 확인용 BED
GATK HaplotypeCaller
GATK의 HaplotypeClaller는 고품질의 변이 검출을 위해 설계되었습니다. 리드에서 발생할 수 있는 시퀀싱 오류를 감지하고 교정하기 위해, 리드의 서브셋을 클러스터링하여 재정렬합니다. 이 과정에서 halpotype을 식별하며, 이는 동일한 DNA 조각에서 유래된 리드의 집합을 의미합니다. 이후, 이 haplotype을 참조 서열과 비교하여 변이를 식별합니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 GATK의 HaplotypeCaller는 입력(input) 데이터로 BAM 파일, Reference sequence FASTA 파일과 Genomic Intervals list 파일을 사용합니다. 출력(output) 데이터로는 비변이 영역에 대한 정보를 포함한 gVCF 파일입니다. GVCF(Genomic VCF) 모드로 실행되어 변이가 없는 위치에 대한 정보도 함께 제공하여 후속 joint genotyping에 활용할 수 있습니다.
주요사항
- > GVCF vs VCF 차이점 - VCF: 변이가 있는 위치만 기록 - GVCF: 모든 위치의 신뢰도 정보 포함 1) 변이 위치: 일반 VCF와 동일 2) 비변이 위치: 참조서열과 동일함에 대한 신뢰도 기록 3) Joint genotyping: 여러 샘플 통합 분석 가능 > 품질 평가 기준 - GQ: ≥30 권장 (99.9% 신뢰도) - DP: 15X 이상 권장 (개체 분석 기준) - VAF: 이형접합 0.3-0.7, 동형접합 >0.8
실행 명령어 예시
$program_dir/gatk HaplotypeCaller -I $INPUT_BAM \ -O $OUTPUT_GVCF \ -R $REFERENCE_GENOME \ -L $GENOMIC_INTERVALS \ -OVI true \ --emit-ref-confidence GVCF
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | File | ref_genome | hg38 | Reference sequence FASTA 파일 경로 | |
| Option | File | genomic_intervals | hg38 | Genomic Intervals list 파일 경로 |
결과
-
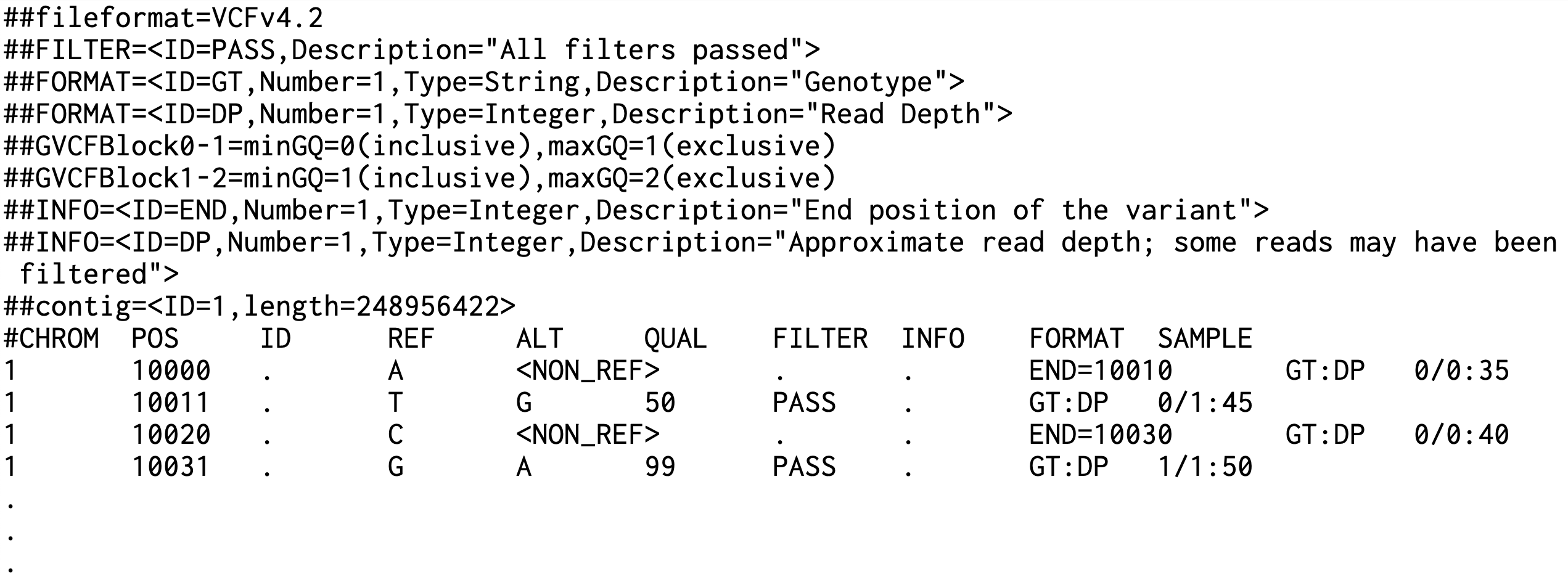
> 파일 헤더 ##fileformat...: VCF 파일 형식 버전 ##ALT...: 특정 위치의 참조 서열과 이미 정의된 대체 서열 외의 모든 가능한 대체 서열 ##FILTER...: 각 샘플에 대한 형식 정보 정의 ##GVCFBlock...: 특정 유전자형 품질 점수 사이의 비변이 영역을 나타내는 gVCF 파일의 블록 정의 ##INFO: 변이 정보에 대한 메타데이터 정의 ##contig: 염색체 또는 컨티그의 이름과 길이 정의 > 데이터 행 #CHROM: 염색체 정보 POS: 위치 정보 ID: 원래 품질 점수변이의 ID REF: 참조 서열 ALT: 대체 서열 또는 ‘<NON_REF>’ (비변이 영역) QUAL: 품질 점수 FILTER: 필터 정보 INFO: 추가 정보 (‘END’ 필드는 비변이 블록의 끝 위치를 나타매) FORMAT: 형식 (‘GT:DP’는 유전자형과 읽기 깊이를 나타냄) SAMPLE: 샘플 정보
BCFtools CountVariants
BCFtools stats는 samtools 프로젝트의 핵심 구성 요소로, VCF/BCF 파일에서 포괄적인 변이 통계를 계산하는 강력한 도구입니다. 이 도구는 생식세포 변이 분석에서 품질 관리와 결과 검증을 위한 필수적인 단계로, GATK HaplotypeCaller에서 생성된 GVCF 파일의 변이 특성을 다각도로 분석하여 시퀀싱 품질과 변이 호출 정확도를 평가합니다. BCFtools stats는 단순한 변이 개수 계산을 넘어서, 유전체학 연구에서 중요한 다양한 지표들을 제공합니다. 이형접합성 비율(heterozygosity ratio), 전이/전환 비율(Ti/Tv ratio), 삽입/결손 분포, 품질 점수 분포, 대립유전자 빈도 스펙트럼 등을 종합적으로 분석하여 샘플의 유전적 특성과 시퀀싱 데이터의 신뢰성을 평가할 수 있습니다. 또한 다중 샘플 분석 시에는 샘플 간 변이 패턴 비교를 통해 배치 효과(batch effect)나 기술적 아티팩트를 감지하는 데도 활용됩니다. 이 도구는 임상 유전체학 분야에서 특히 중요한데, 변이 호출 결과의 품질을 객관적으로 평가하여 후속 해석 과정의 신뢰성을 보장하는 역할을 합니다. 예를 들어, 예상 범위를 벗어나는 Ti/Tv 비율이나 비정상적인 이형접합성 패턴은 샘플 오염, 시퀀싱 오류, 또는 분석 파이프라인의 문제를 시사할 수 있어 이러한 지표들을 통한 체계적인 품질 관리가 필수적입니다.
주요사항
- [품질 문제 진단 가이드] 1. Ti/Tv 비율 이상 > 낮은 Ti/Tv (<2.0 WGS, <3.0 WES): - 원인: 과도한 transversion 호출, 낮은 품질 변이 포함 - 해결: 품질 필터 강화 (QUAL ≥ 30, GQ ≥ 30) > 높은 Ti/Tv (>2.2 WGS, >3.6 WES): - 원인: transition 편향 호출, 특정 서열 컨텍스트 편향 - 해결: 정렬 품질 확인, 참조 유전체 버전 점검 2. 이형접합성 이상 > 과도한 이형접합 (Het/Hom >3.0): - 원인: 샘플 오염, 대립유전자 dropout - 해결: ContEST, VerifyBamID2로 오염도 확인 > 과도한 동형접합 (Het/Hom <1.0): - 원인: 근친혼, 염색체 이상, LOH - 해결: ROH 분석, 염색체 이상 검사 3. 변이 개수 이상 > 변이 과다 (>120% 기준값): - 원인: 샘플 오염, 낮은 품질 기준 - 해결: 품질 필터 강화, 오염도 검사 > 변이 부족 (<80% 기준값): - 원인: 과도한 필터링, 낮은 커버리지 - 해결: 필터 기준 완화, 시퀀싱 깊이 확인
실행 명령어 예시
$PROGRAM_DIR/bcftools stats -s - $INPUT_VCF | grep PSC
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | 분석할 BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 |
결과
-
.png)
[sample]_countVariants.txt : 변이 통계 결과 정리 파일 [4] nRefHom: 참조 동형접합 SNP 수 [5] nNonRefHom: 변이 동형접합 SNP 수 [6] nHets: 이형접합 SNP 수 [7] nTransitions: 전이 변이 수 (A↔G, C↔T) [8] nTransversions: 전환 변이 수 (A↔C, A↔T, G↔C, G↔T) [9] nIndels: 삽입/결손 변이 수 [10] average depth: 평균 시퀀싱 깊이 [11] nSingletons: 단일 샘플에만 나타나는 변이 수 [14] nMissing: 결측 유전형 수
Manta Single-Diploid
Manta Single-Diploid는 개체 시퀀싱 데이터에서 생식세포 구조 변이(germline structural variants)를 검출하는 도구입니다. 결손(deletion), 삽입(insertion), 역위(inversion), 전좌(translocation), 중복(duplication) 등 50bp 이상의 큰 규모 유전체 구조 변화를 찾아냅니다. 이 도구는 split-read 분석을 통해 구조 변이 경계를 정확히 매핑하고, discordant read-pair 분석으로 비정상적 insert size를 가진 구조 변이를 감지합니다. 복잡한 구조 변이 영역에서는 local assembly 기법을 사용하여 정확한 재구성을 수행하며, multi-threading을 지원하여 병렬 처리를 통한 빠른 분석 속도를 제공합니다. 특히 낮은 coverage 환경에서도 높은 민감도로 구조 변이를 검출할 수 있어 다양한 시퀀싱 프로젝트에 적용 가능합니다. Manta는 BWA, Bowtie2 등으로 정렬된 BAM 파일과 압축된 CRAM 파일을 모두 지원하며, paired-end와 single-end 데이터 형식 모두에서 작동합니다. 또한 단일 샘플 분석뿐만 아니라 동시에 여러 샘플을 분석할 수 있는 기능을 제공하여 대규모 코호트 연구에도 효율적으로 활용할 수 있습니다.
주요사항
- > 품질 필터링 기준 1. PASS 필터 조건 - 최소 지지 리드: Split-read ≥ 3, Paired-read ≥ 3 - Genotype Quality: GQ ≥ 15 - Mapping Quality: 매핑된 리드의 평균 MAPQ ≥ 20 - Repeat Content: 반복 서열 영역 회피 - Assembly Quality: Local assembly 성공 2. 필터 태그 의미 - PASS: 모든 품질 기준 통과 - LowGQ: 낮은 유전형 품질 (GQ < 15) - LowGQX: 확장 품질 점수 부족 - HomRef: 참조 동형접합 (변이 없음) - NotGenotyped: 유전형 결정 실패
- > Manta 결과의 복잡한 역위(inversion) 변이의 convertInversion.py 처리를 통한 표준화된 형태로의 변환 전후 비교 예시 - 변환 전 (복잡한 형태): chr3 100000 . N ]chr3:200000]N . PASS SVTYPE=BND;MATEID=chr3_100000_2 chr3 200000 . N N[chr3:100000[ . PASS SVTYPE=BND;MATEID=chr3_100000_1 - 변환 후 (표준화): chr3 100000 . N <INV> . PASS SVTYPE=INV;SVLEN=100000;END=200000 - 원본 vs 변환후 구조 변이 수 비교 Original_INV: 8 complex_breakpoints Converted_INV: 12 standard_inversions 전체적으로 SV 4개 증가: 복잡한 BND가 단순한 INV로 병합됨
- > 구조 변이 유형별 특징
- 1. DEL (Deletion) - 결손 - 크기 범위: 50bp - 수 Mb - 검출 방법: Split-read + Discordant pair - 임상 의미: 유전자 전체 또는 일부 소실 - 예상 빈도: 개체당 약 4,000-6,000개
- 2. DUP (Duplication) - 중복 - 유형: Tandem duplication, Interspersed duplication - 크기 범위: 50bp - 수 Mb - 검출 방법: Read depth + Discordant pair - 임상 의미: 유전자 복제수 증가 - 예상 빈도: 개체당 약 500-1,000개
- 3. INV (Inversion) - 역위 - 유형: Paracentric, Pericentric - 크기 범위: 1kb - 수 Mb - 검출 방법: Split-read cluster 분석 - 임상 의미: 유전자 발현 또는 기능 변화 - 예상 빈도: 개체당 약 100-300개
- 4. TRA (Translocation) - 전좌 - 유형: Balanced, Unbalanced - 검출 방법: Discordant read-pair 분석 - 임상 의미: 염색체간 재배열 - 예상 빈도: 개체당 약 50-200개
- 5. INS (Insertion) - 삽입 - 유형: Mobile element, Novel sequence - 크기 범위: 50bp - 수 kb - 검출 방법: Split-read + Assembly - 임상 의미: 새로운 서열 삽입 - 예상 빈도: 개체당 약 500-1,500개
실행 명령어 예시
#1. Workflow Setup: Generate configuration files and working scripts for analysis python $PROGRAM_DIR/bin/configManta.py \ --bam $INPUT_BAM \ --referenceFasta $ref_genome \ --runDir $SAMPLE_output_dir #2. Workflow Execution: Execute the configured workflow to perform actual SV detection python $SAMPLE_output_dir/runWorkflow.py \ -m local –j 6 #3. Convert INV represented by two breakends into a single INV record to enhance interpretability python $PROGRAM_DIR/libexec/convertInversion.py \ $PROGRAM_SAMTOOLS \
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | BAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | File | ref_genome | hg38 | Reference sequence FASTA 파일 경로 |
결과
-
.png)
> Manta Single-Diploid 결과 파일들의 구조 및 설명
-
.png)
> 중요 VCF 필드 해석 SVTYPE: 구조 변이 유형 (DEL/DUP/INV/TRA/INS) SVLEN: 변이 크기 (음수: 결손, 양수: 삽입) CIPOS/CIEND: 위치 신뢰구간 (breakpoint uncertainty) PR: Paired-read 지지도 (참조:변이) SR: Split-read 지지도 (참조:변이) GQ: 유전형 품질 점수