- 카테고리 Epigenomics > DNA-binding-protein-based-analysis
- 수정일2025-10-31 17:20:24
- 레퍼런스
Bio-Express ChIP-seq Analysis Pipeline은 크로마틴 면역침전 시퀀싱(Chromatin Immunoprecipitation Sequencing) 데이터로부터 단백질-DNA 결합 부위를 검출하기 위한 모듈식 분석 파이프라인입니다. 이 파이프라인은 raw FASTQ 파일을 입력으로 사용하고, 전사인자 결합 사이트, 히스톤 변형 영역, 크로마틴 구조 분석을 기반으로 하는 포괄적인 후성유전학적 결합 부위 호출 결과와 품질 평가 및 시각화를 제공합니다. FastQC를 통한 시퀀싱 품질 평가 후, FASTX-Toolkit을 사용하여 저품질 염기 필터링을 진행하고, Bowtie2 정렬 도구를 사용하여 참조 유전체 서열에 매핑하여 SAM 형식의 정렬 파일을 생성합니다. 이후 전처리가 완료된 정렬 파일을 활용하여 후성유전학적 신호 분석 단계로 진입합니다. MACS2(Model-based Analysis of ChIP-Seq)를 통한 통계적으로 유의한 피크 호출을 수행하여 단백질-DNA 결합 부위를 정확히 식별하고, narrowPeak 형식으로 고해상도 결합 영역을 제공합니다. 최종적으로 Homer를 활용한 포괄적인 후속 분석 단계를 수행합니다. annotatePeaks 기능을 통해 검출된 피크의 게놈 위치 주석과 주변 유전자 정보를 제공하고, makeUCSCfile을 사용하여 UCSC 게놈 브라우저와 호환되는 bedGraph 형식의 시각화 파일을 생성하여 크로마틴 면역침전 신호의 게놈 전체 분포 패턴을 직관적으로 확인할 수 있습니다.
> 기본 참조 게놈: hg38
[중요] 샘플 유형 식별 방법:
- 컨트롤 파일: "CONTROL_"로 시작 필수 (자동 식별을 위한 필수 접두사)
- 처리/ChIP 파일: 특별한 파일명 규칙 없음
(예시)
CONTROL_input_R1.fastq.gz # 유효한 컨트롤, Read 1
CONTROL_input_R2.fastq.gz # 유효한 컨트롤, Read 2
ChIP_H3K4me3_R1.fastq.gz # 유효한 처리군, Read 1
ChIP_H3K4me3_R2.fastq.gz # 유효한 처리군, Read 2
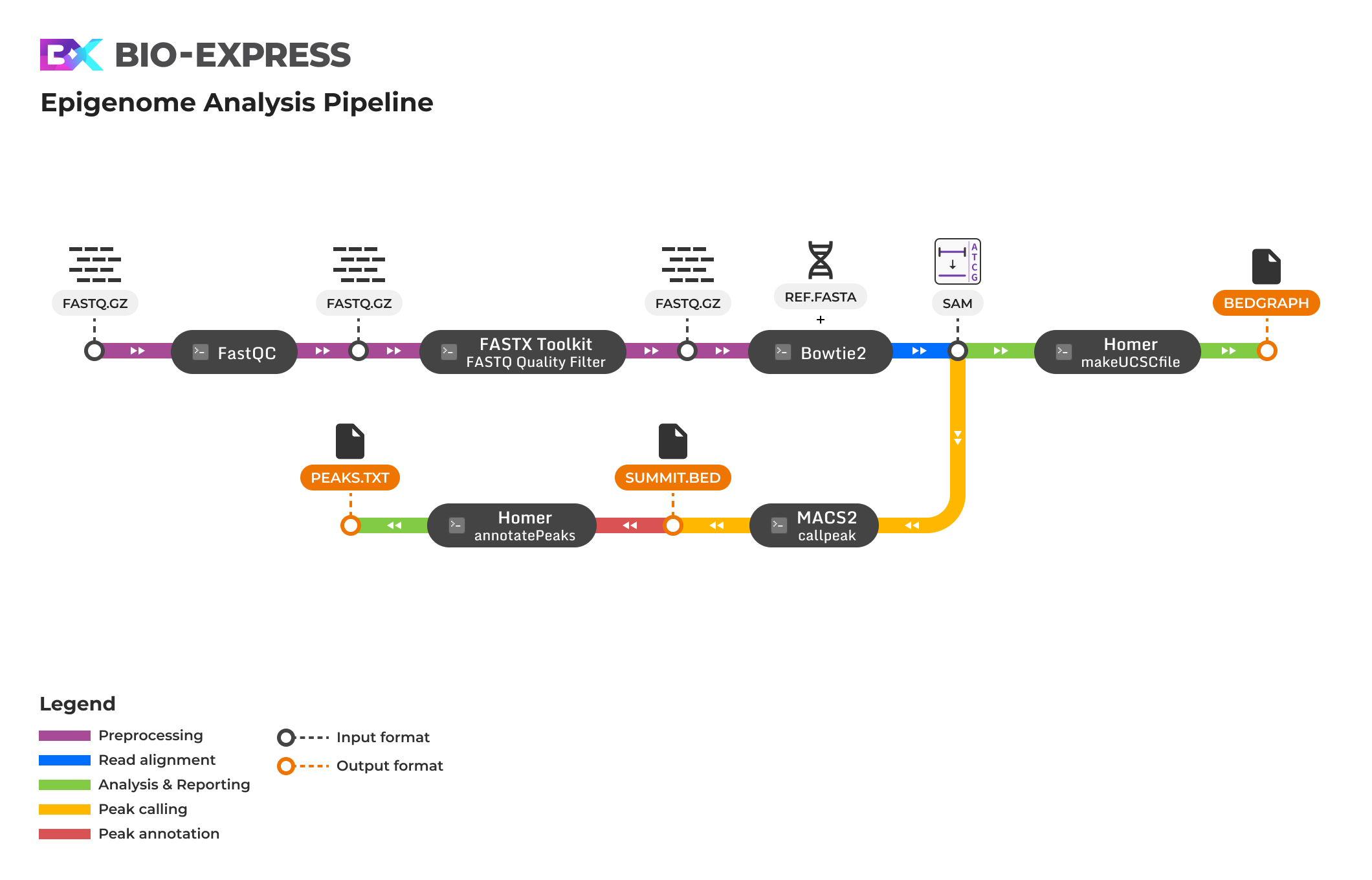
파이프라인 모듈
FastQC
FastQC는 고속 염기서열 분석(high throughput sequence) 데이터의 품질 관리를 위한 분석도구입니다. 이 프로그램은 FASTQ 형식의 서열 데이터를 읽어들여 여러 품질 관리(Qaulity Control) 검사를 수행하고 결과는 HTML 기반의 보고서로 출력합니다. FastQC는 전반적인 품질 문제에 대한 개요 정보를 제공하며, 쉽게 확인할 수 있는 요약된 그래프와 테이블을 포함합니다. FastQC는 FASTQ 형식의 파일이 입력 파일로 사용되며, 출력 결과는 리포트 html 파일과 zip 형식의 압축 파일이 생성됩니다.
주요사항
- FastQC는 자바 애플리케이션입니다. 실행하기 위해서는 시스템에 적절한 자바 실행 환경(Java Runtime Environment, JRE)이 설치되어 있어야 합니다. 따라서 FastQC를 실행하기 전에 먼저 적절한 JRE가 설치되어 있는지 확인해야 합니다. 다양한 종류의 JRE를 사용할 수 있지만, 저희가 테스트해본 것은 최신 오라클 런타임 환경과 adoptOpenJDK 프로젝트의 JRE입니다. 64비트 JRE를 다운로드하여 설치하고, 자바 애플리케이션이 시스템 경로(path)에 포함되도록 설정해야 합니다(대부분의 설치 프로그램이 이를 자동으로 처리해줍니다).
실행 명령어 예시
$program_dir/fastqc –t 6 –o $OUTPUT_DIR $INPUT_DIR/$READ
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o) |
결과
-
.png)
Basic Statistics 테이블은 주어진 FASTQ 파일에 대한 간단한 통계적 정보를 제공합니다. 일반적으로 다음과 같은 정보를 포함합니다. Filename : 분석된 파일의 이름 또는 경로 File type : FASTQ 파일의 종류 Encoding : 품질 점수 인코딩 방식 Total Sequence : 총 서열 수 Filtered Sequences : Read 품질이 좋지 않은 서열 수 Sequence length : 서열의 길이 %GC : 서열에서의 GC 백분율
-
.png)
X축은 리드의 염기 위치를 나타내며, Y축은 품질 점수를 의미합니다. 점수가 높을수록 품질이 좋습니다. 중앙의 빨간색 선은 중앙값을 나타내고, 노란색 박스는 사분위간 범위(25~75%)를 의미합니다. 위쪽 및 아래쪽의 위스커(whisker)는 각각 10% 및 90% 포인트를 나타냅니다. 파란색 선은 평균 품질을 의미합니다. 어떤 염기의 하위 사분위수가 10 미만이거나 중위값이 25보다 작을 경우 경고로 간주됩니다. 또한, 어떤 염기의 하위 사분위수가 5 미만이거나 중위값이 20보다 작으면 오류로 간주됩니다.
-
.png)
색을 이용하여 각 타일의 품질을 나타내며, 파란색은 품질이 높음을 나타내고 빨간색은 품질이 낮음을 나타냅니다. 각 타일의 품질을 모든 타일의 평균 품질과 비교하여 예상 패턴과의 편차를 식별 할 수 있습니다. 특정 타일의 품질이 지속적으로 좋지 않으면 물리적 결함이나 오염 등 셀의 특정 영역에 문제가 있음을 나타낼 수 있습니다. 이상적으로는 모든 타일이 높은 품질을 보여야 하면 플롯에서 더 차가운 색상으로 표시됩니다. 이 플롯은 Illumina 라이브러리에서만 나타납니다.
-
.png)
X축은 리드의 전체 길이에 대한 평균 품질 점수를 나타내고, Y축은 해당 품질 점수를 갖는 읽기의 수를 나타냅니다. 시퀀스의 시퀀싱 품질은 해당 시퀀스에 대한 정확성과 신뢰성을 나타내며, 품질 점수가 높을수록 오류 발생 가능성이 낮다는 것을 의미합니다. 만약 시퀀싱 실행이 전반적으로 낮은 품질을 보인다며, 시퀀싱 화학 문제나 샘플 준비 문제 등이 있을 수 있습니다. 품질 점수가 기록되지 않은 BAM/SAM 파일의 경우 확인할 수 없습니다. 품질 점수가 Phred 척도 기준 최고 품질 점수 27점(오류율 0.2%) 미만일 경우 경고 발생, 20점(오류율 1%) 미만일 경우 오류입니다.
-
.png)
X축은 리드의 포지션을 나타내고, Y축은 시퀀싱한 리드에서 각 base의 전체 비율을 나타냅니다. 좋은 품질의 시퀀싱 샘플에서는 각 위치의 염기 비율을 나타내는 4개의 선이 평행하고 서로 가까워야 합니다. 그러나 선이 일부 위치에서 엉키거나 얽히면 과도하게 표현된 시퀀스가 오염되었음을 나타낼 수 있습니다. 또한, A/T 또는 G/C 염기의 비율이 어떤 위치에서는 10% 이상 차이나면 경고 발생, 20%를 초과하면 오류입니다.
-
.png)
시퀀스에서 G와 C 뉴클레오티드의 백분율 비율을 나타냅니다. 이를 통해 DNA 또는 RNA 시퀀스의 특성을 이해할 수 있습니다. 시퀀스의 GC 함량은 DNA 안정성, 서열의 물리적 특성, 유전자 발현에 영향을 미칠 수 있으므로 중요한 지표 중 하나입니다. X축은 GC contents의 비율을 나타내고, Y축은 시퀀스의 총량을 나타냅니다. 정규 분포와 편차 합계가 전체 리드의 15%를 초과하면 경고, 30%를 초과하면 오류입니다.
-
.png)
시퀀싱 리드의 각 위치에서 발견된 N 비율을 나타내며 일반적으로 매우 낮습니다. 그러나 어떤 위치에서는 N 비율이 5%를 초과하면 시퀀싱 시스템에 문제가 있을 수 있다는 경고로 간주되며, 20%를 초과하면 오류로 간주됩니다. 데이터의 품질이 높은지 확인하고 시퀀싱 읽기의 정확성에 영향을 줄 수 있는 문제를 식별하려면 시퀀싱 중에 N 비율을 모니터링하는 것이 중요합니다. N 비율이 권장 임계값을 초과하는 경우 문제의 심층적인 분석이 필요할 수 있습니다.
-
.png)
시퀀싱 데이터에서 각 리드의 길이에 대한 분포를 나타냅니다. 이 그래프의 X 축은 시퀀스 길이를, Y 축은 리드 수를 나타냅니다. 시퀀싱 데이터에서 발견된 리드의 길이가 어떻게 분포되어 있는지를 시각적으로 확인할 수 있습니다. 이 그래프를 통해 시퀀싱 데이터 세트의 리드 길이 분포를 파악할 수 있으며, 예상치 못한 리드 길이나 이상한 분포를 감지하여 데이터의 품질을 평가하는 데 도움이 됩니다. 일반적으로 시퀀스 길이 분포는 일정하거나 특정한 패턴을 따르지만, 비정상적인 분포는 시퀀싱 데이터에 문제가 있을 수 있다는 신호일 수 있습니다.
-
.png)
중복된 시퀀스가 전체의 20% 이상일 경우 경고, 50% 이상일 경우 오류입니다.
-
.png)
시퀀싱 데이터에서 빈번하게 등장하는 시퀀스를 나타내는 테이블입니다. 이 테이블은 시퀀싱 데이터에서 특정 시퀀스가 기대보다 더 자주 나타나는 경우를 식별합니다. 실험과정에서 발생한 오류, PCR 이중성, 어댑터 오류 또는 샘플의 비정상적인 특정으로 인해 발생할 수 있습니다. 해당 내용을 확인하고 잠재적인 문제를 식별하는 것은 시퀀싱 데이터의 정확성과 신뢰성을 높이는 데 도움이 됩니다.
-
.png)
시퀀싱 데이터에서 발견된 어댑터 시퀀스의 누적 백분율을 보여주는 차트입니다. 시퀀스가 발견된 위치에 따라 백분율이 증가하며, 시퀀스가 리드의 끝까지 존재하는 동안 계산됩니다. 어댑터의 비율을 확인하여 데이터의 품질을 평가하며, 어댑터 시퀀스가 발견되는 비율이 높을수록 시퀀싱 데이터에 오류가 포함될 가능성이 높아지므로, 5%를 초과하면 경고, 10%를 초과하면 오류입니다.
Cutadapt
Cutadapt는 NGS 시퀀싱 데이터에서 어댑터(adapter) 서열 제거, 품질이 낮은 서열 제거 등 다양한 전처리 작업을 수행하는 데 사용되는 도구입니다. 주로 서열의 3’ 말단에서 염기서열을 제거하며, 특히 RNA 시퀀싱 또는 DNA 시퀀싱 데이터에서 어댑터 제거에 빈번하게 활용됩니다. 전체 데이터의 품질을 유지하여 이후의 분석 단계에서 정확한 결과를 얻을 수 있도록 도움을 줍니다. 입력 데이터로는 fastq, fastq.gz, fq, fq.gz 확장자를 가진 파일을 사용할 수 있습니다. 출력으로는 파일 확장자 앞쪽에 ‘_trimmed’라는 문구가 붙은 fastq 파일이 생성되도록 세팅되어 있습니다. 따라서 "trimmed.fastq" 파일은 프라이머 잔여물이나 다른 불순물이 제거된 reads 파일을 나타냅니다.
주요사항
- 1. Bio-Express의 Cutadapt 모듈은 paired-end와 single-end 형식의 FASTQ 데이터를 모두 처리할 수 있도록 설계되었습니다.
- 2. -a 및 –A 옵션에 입력해야 할 각각의 어댑터 서열은 NGS 시퀀서 플랫폼별로 다음과 같습니다. > Illumina: 기본값 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT > MGI: 필요시 기본값 대체 입력 -a AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA -A AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG
실행 명령어 예시
$PROGRAM_DIR/cutadapt -q 20 –Q 20 -j 6 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 --pair-filter=$PAIR_FILTER -o $OUTPUT_DIR/$READ_1 –p $OUTPUT_DIR/$READ_2 $INPUT_DIR/$READ_1 $INPUT_DIR/$READ_2
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o, -p) | |
| Option | Integer | min_len_r1 | 70 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read1의 최소 길이 | |
| Option | Integer | min_len_r2 | 1 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read2의 최소 길이 | |
| Option | String | pair_filter | any | --pair-filter (single-end FASTQ에는 미적용) - any: 두 Read 중 어느 하나라도 조건에 부합하면 필터링 - both: 두 Read 모두 조건에 부합하면 필터링 | |
| Option | String | adapter_pos | adapter | 어댑터 시퀀스 처리 위치 지정 --adapter: 서열의 3' 끝 방향에서 어댑터를 찾아 해당 어댑터와 그 이후 모든 서열을 제거 (-a, -A) --front: 서열의 5' 시작 방향에서 어댑터를 찾아 해당 어댑터와 그 이전 모든 서열을 제거 (-g, -G) --anywhere: 5' 또는 3' 어느 쪽에든 나타날 수 있는 어댑터를 탐지 (-b, -B) | |
| Option | String | adapter_r1 | AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | 첫 번째 Read의 어댑터 시퀀스 (-a) | |
| Option | String | adapter_r2 | AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT | 두 번째 Read의 어댑터 시퀀스 (-A) | |
| Option | Integer | quality_r1 | 20 | 첫 번째 Read의 절단 기준으로 사용할 품질 임계값 (-q) | |
| Option | Integer | quality_r2 | 20 | 두 번째 Read의 절단 기준으로 사용할 품질 임계값 (-Q) |
결과
-
.png)
총 입력된 read pair 수, 어댑터가 검출된 read의 수와 비율, 필터링 후 최종적으로 출력된 read 수 등 전체 리드 처리 현황을 요약함. 또한 데이터 전처리 단계에서 몇 %의 리드가 어댑터에 의해 잘렸고, 몇 %가 최종 분석에 사용 가능한지, 데이터 손실이 얼마나 발생했는지 파악함.
-
.png)
어댑터 서열 (Illumina adapter 등), 탐지 및 제거된 횟수, 최소 overlap 길이, mismatch 허용 개수 (error tolerance), 제거된 어댑터 서열에서의 염기 비율(어댑터 클리핑의 정확성 평가에 활용) 등 Read1에서 탐지된 어댑터의 정보. Read2에 대한 내용도 동일한 방식으로 출력함.
Bowtie2
Bowtie2는 Johns Hopkins University의 Ben Langmead와 Steven Salzberg가 개발한 플랫폼으로, 전 세계 주요 게놈 센터와 바이오인포매틱스 분야에서 서열 정렬 분야의 표준 참조로 인정받고 있는 핵심 도구입니다. 이 도구는 기존 정렬 도구들의 한계를 극복하기 위해 FM-index(Full-text Minute-space index) 기술과 BWT(Burrows-Wheeler Transform) 알고리즘을 결합한 획기적인 색인 구조를 구현하여, 인간 전체 게놈과 같은 거대한 참조 서열에서도 메모리 효율성과 검색 속도를 동시에 최적화한 혁신적 솔루션을 제공합니다. Bowtie2의 가장 큰 기술적 혁신은 갭을 허용하는 정렬(gapped alignment)과 지역 정렬(local alignment) 기능을 완벽하게 지원한다는 점입니다. 이는 ChIP-seq 분석에서 핵심적으로 중요한데, 크로마틴 면역침전 과정에서 발생할 수 있는 DNA 단편화나 시퀀싱 오류를 지능적으로 처리하면서도 multi-mapping 리드의 정확한 분류를 통해 반복 서열 영역에서의 false positive 신호를 효과적으로 차단합니다. 특히 MAPQ(Mapping Quality) 점수 계산 시스템을 통해 각 정렬의 신뢰도를 정량적으로 평가하여, 후속 MACS2 피크 호출에서 고유하게 매핑된 고신뢰도 리드만을 선별적으로 활용할 수 있게 합니다.
주요사항
- $input_dir에는 Single-end와 paired-end FASTQ 파일 모두 처리 가능합니다. 이는 fastq 또는 fq 확장자 앞에 있는 “_R1/2”, “.R1/2”, “_1/2” 형식의 존재 여부로 single 및 paired end 여부를 자동으로 처리합니다.
- $reference_genome에는 샘플이 해당하는 종의 참조 서열의 FASTA 파일을 입력합니다. FASTA 파일 입력 시 자동으로 bowtie2 mapping에 필요한 index 파일을 생성합니다. 미리 파일이 생성된 경우 이 과정은 생략됩니다.
실행 명령어 예시
/opt/apps/Bowtie2/bowtie2 \ -x /path/to/reference_genome_file/[bowite2_index_prefix] \ -1 /path/to/input_dir/[name]_R1.fastq.gz \ -2 /path/to/input_dir/[name]_R2.fastq.gz \ -S /path/to/output_dir/[name].sam
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | /path/to/input_dir | FASTQ 파일이 있는 경로 | |
| Output | Folder | output_dir | /path/to/output_dir | Reference genome FASTA에 FASTQ 데이터가 매핑되어 생성된 SAM 파일이 생성될 경로 | |
| Option | File | ref_genome | hg38 | Reference genome FASTA 파일. Bowtie2 index의 basename. |
결과
-
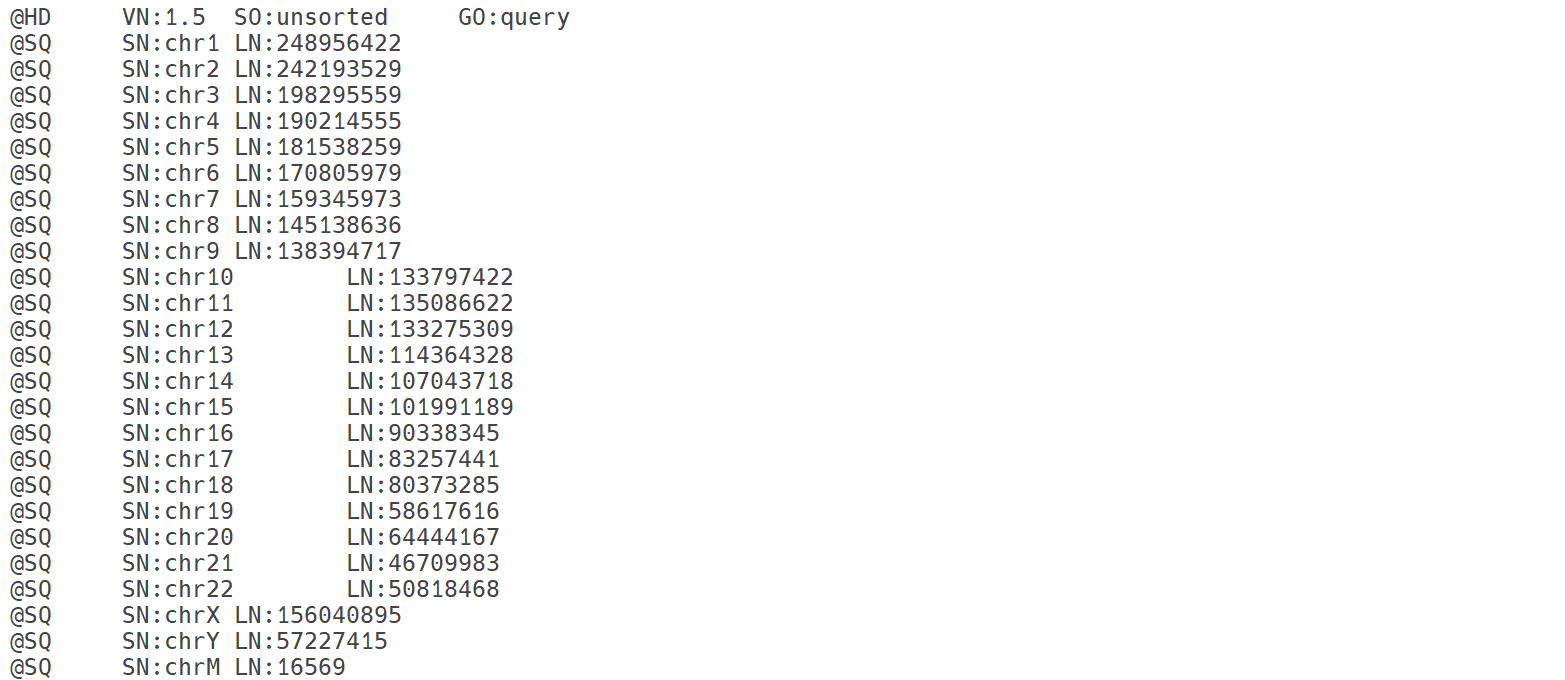
SAM 파일의 header 영역. @HD: header 영역 전체를 정의. VN: SAM 파일의 포맷 버전 SO: 정렬 방식 GO: 그룹화 방식. query는 read 이름 별로 그룹화되었음을 의미. @SQ: 참조 서열에 대한 정보를 제공함. 이 경우 사람 genome의 각 염색체에 대한 정보를 제공함. @RG: read 그룹의 정보를 제공함. @PG: 프로그램 그룹의 정보로, 정렬 도구 등의 정보를 제공함.
-
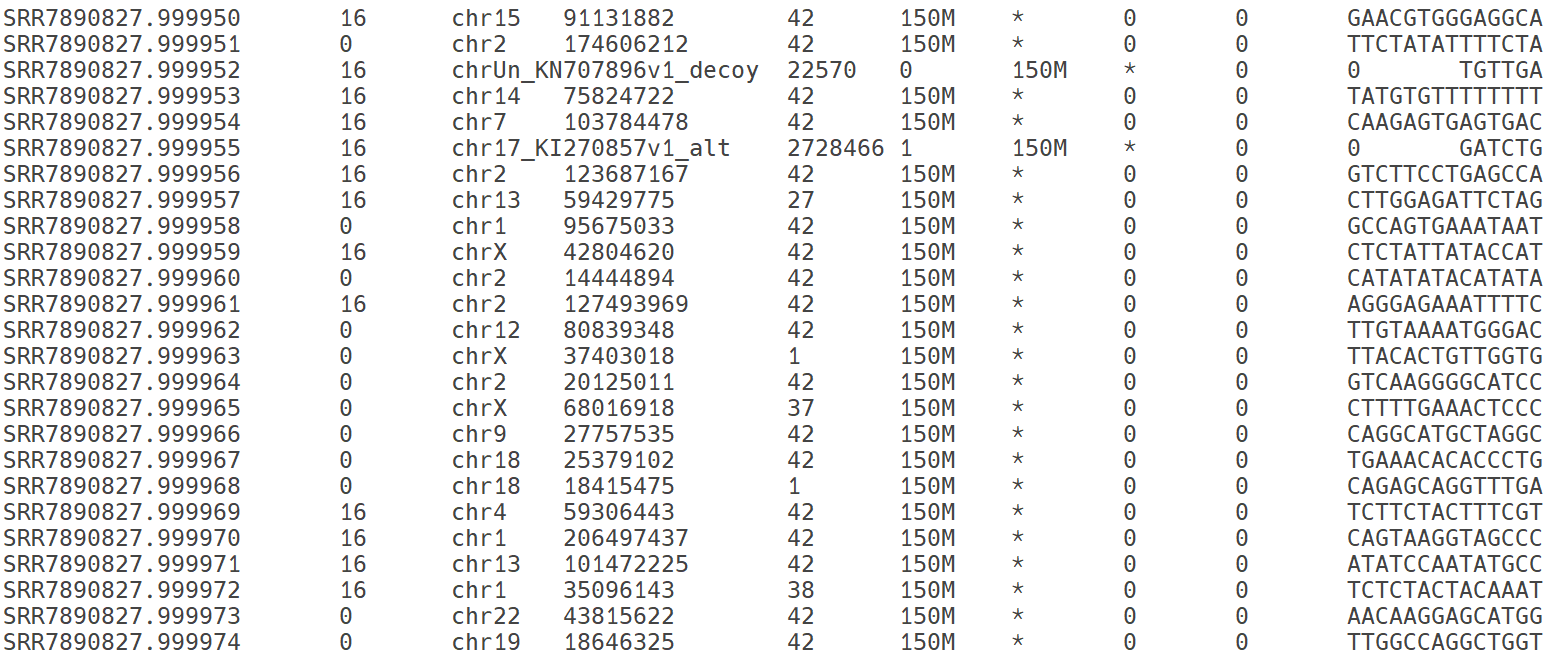
SAM 파일의 본문 영역. mapping 정보를 제공. 제시된 이미지의 각 열은 다음과 같은 정보를 제공함: read 이름, 정렬 상태, 참조서열 이름, 정렬의 왼쪽 위치, 정렬 품질, 정렬 상태, paried-end의 다음 read 정렬 정보, paried-end의 두 read 사이의 거리, read의 염기서열 정보를 제공하고 있음.
Homer_makeucscfiles
Homer makeUCSCfile은 University of California San Diego(UCSD)의 Homer 생태계 내에서 ChIP-seq 데이터 시각화 분야의 혁신적 솔루션으로 개발된 고성능 데이터 변환 엔진으로, UCSC Genome Browser, Ensembl, IGV 등 주요 게놈 브라우저와의 완벽한 호환성을 제공하는 표준 시각화 파이프라인입니다. 이 도구는 단순한 파일 형식 변환을 넘어서 정교한 신호 정규화 알고리즘과 다중 해상도 시각화 최적화 기술을 통합하여, 거대한 ChIP-seq 데이터셋을 실시간 브라우징이 가능한 효율적인 형태로 변환하면서도 생물학적 신호의 정량적 정확성을 완벽하게 보존합니다. Homer makeUCSCfile의 핵심 혁신은 태그 디렉토리 기반 데이터 구조화 시스템에 있습니다. 이 도구는 원시 정렬 파일(SAM/BAM)을 염색체별로 분할된 효율적인 태그 디렉토리 구조로 재구성하여 메모리 사용량을 최소화하면서도 빠른 랜덤 액세스를 가능하게 하고, 다양한 정규화 방법(RPM, RPKM, TPM 등)을 적용하여 샘플 간 비교 가능한 표준화된 신호 강도를 생성합니다. 특히 fragment length 보정 알고리즘을 통해 ChIP-seq 실험의 단편 크기 분포를 정확하게 반영하고, strand-specific signal processing으로 방향성 있는 단백질 결합 패턴을 정밀하게 시각화합니다. Homer makeUCSCfile은 bedGraph와 bigWig 포맷의 이중 지원을 통해 사용자의 다양한 분석 요구를 충족시키며, 컨트롤 샘플 기반 배경 제거 기능으로 IP-specific 신호만을 선별적으로 강조하고, multi-scale visualization optimization을 통해 전체 게놈 뷰에서 단일 염기 해상도까지의 모든 줌 레벨에서 최적화된 시각적 표현을 제공합니다. 이러한 포괄적 시각화 솔루션을 통해 연구자들은 복잡한 ChIP-seq 데이터를 직관적이고 정확한 게놈 브라우저 트랙으로 변환하여 가설 생성, 패턴 발견, 결과 검증, 그리고 연구 결과 발표에 필수적인 고품질 시각적 증거를 생성할 수 있습니다. 입력 데이터는 Bowtie2가 완료된 aligned.sam 파일이며, ChIP-Seq 데이터의 전처리 결과인 각 분석 샘플의 염색체별 tags.tsv 파일, 기본 태그 정보와 시퀀싱 실행에 관한 내용이 적혀 있는 tagInfo.txt 파일, read mapping 결과를 UCSC Genome Browser 및 IGV에서 시각화할 수 있는 BedGraph 및 BigWig 형식의 파일 등으로 구성된 출력물이 생성됩니다.
주요사항
- > UCSC Genome Browser를 이용한 분석 결과 확인 1. UCSC Genome Browser 접속 2. "My Data" → "Custom Tracks" 선택 3. 확인하고자 하는 Genome 종류 및 Assembly 버전 선택 4. '파일 선택' 버튼 클릭 후, bedGraph 데이터 선택 5. 'Submit' 버튼 클릭하여 업로드 6. Manage Custom Tracks 항목의 Name 링크 클릭 7. 아래와 같은 configuration 정보 확인 및 수정 track type=bedGraph \ name="sample_ChIP" \ description="ChIP-seq signal bedGraph" \ visibility=full \ autoScale=on \ alwaysZero=on \ viewLimits=0:10 \ color=200,100,0 \ priority=20 > UCSC Genome Browser 상세 설정 옵션 - visibility: 표시 모드 (hide/dense/pack/squish/full) - autoScale: 자동 스케일링 활성화 - alwaysZero: Y축 0부터 시작 - viewLimits: 표시 범위 지정 (최소값:최대값) - color: RGB 색상 값 (예: 200,100,0 = 주황색) - priority: 트랙 표시 순서 > IGV(Integrative Genomics Viewer)를 이용한 분석 결과 확인 - 로컬 파일 로딩 1. File → Load from File 선택 2. BedGraph 파일(.bedGraph 또는 .bg) 선택 = IGV 표시 옵션 설정 - 우클릭 → Set Data Range: 표시 범위 수동 설정 - Auto-scale: 자동 스케일링 옵션 - Log Scale: 로그 변환 표시 (높은 신호값 데이터용) - Color: 트랙 색상 변경
실행 명령어 예시
/opt/apps/HOMER/makeTagDirectory /path/to/output_dir /path/to/input_dir/[name].sam /opt/apps/HOMER/makeUCSCfile /path/to/output_dir -o [name].BedGraph \ /path/to/ugbtools_dir/bedGraphToBigWig [name].BedGraph hs18 [name].BigWig
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | .SAM 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 |
결과
-
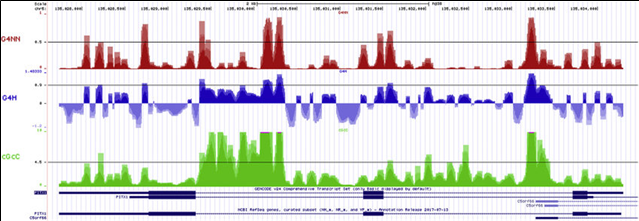
UCSC Genome Browser에서 확인한 bedGraph 파일
-

[sample_name]|[chr_name].tags.tsv chr1: 염색체 이름 14943: Peak의 시작 위치 또는 현재 위치 (1/0): Peak의 양성 여부. 1은 Peakk가 양성. 0은 Peak가 음성 1.0: Peak의 신뢰도 또는 품질을 의미 100: Peak의 점수 또는 품질을 의미
-
.png)
[sample_name]/tagInfo.txt name: dataset 또는 염색체의 이름 Unique Positions: 해당 dataset 또는 염색체에서 고유한 위치(1389825) Total Tags: 해당 dataset 또는 염색체에서 전체 태그 수(1396047.0) fragmentLengthEstimate: DNA 단편의 길이 추정치 peakSizeEstimate: Peak의 크기 추정치 tagsPerBP: 각 염색체 위치당 평균 태그 수 averageTagsPerPosition: 각 위치에 대한 평균 태그 수 medianTagsPerPosition: 각 위치에 대한 중앙값 태그 수 averageTagLength: 평균 태그의 길이 gsizeEstimate: 게놈 크기의 추정치 averageFragmentGCcontent: 평균 DNA 단편의 GC 함유량
MACS2_callpeak
MACS2 (Model-based Analysis of ChIP-Seq)는 Harvard University의 Xiaole Shirley Liu Lab에서 개발한 ChIP-seq 피크 호출 분야의 절대적 표준(absolute standard)으로, ENCODE 프로젝트, NIH Roadmap Epigenomics, 그리고 전 세계 수천 개의 연구기관에서 크로마틴 면역침전 데이터 분석의 필수 도구로 채택되어 10,000편 이상의 논문에서 인용된 검증된 플랫폼입니다. 이 도구는 기존의 단순한 임계값 기반 방법들의 한계를 극복하기 위해 베이지안 통계학적 프레임워크와 포아송 분포 기반 확률 모델을 통합한 혁신적인 알고리즘을 구현하여, ChIP-seq 실험의 생물학적 변이성과 기술적 노이즈를 동시에 고려한 정교한 통계적 검정을 수행합니다. MACS2의 핵심 혁신은 동적 람다(dynamic lambda) 모델링 시스템에 있습니다. 이 도구는 게놈의 각 영역에서 국지적 배경 분포를 독립적으로 추정하여(1kb, 5kb, 10kb, 전체 게놈의 4단계 스케일), 크로마틴 접근성 차이, GC 함량 편향, 반복 서열 밀도 등에 의한 지역별 배경 신호 변동을 정밀하게 보정합니다. 특히 shift size 추정 알고리즘을 통해 ChIP-seq 실험 특유의 단편 크기와 스트랜드 편향 패턴을 자동으로 감지하고, cross-correlation 분석을 기반으로 최적의 태그 확장 매개변수를 실험별로 맞춤 설정합니다. 더 나아가 MACS2는 Benjamini-Hochberg FDR 보정과 q-value 계산을 통해 다중 검정 문제를 엄격하게 해결하고, broad peak 모드에서는 히스톤 변형과 같은 넓은 크로마틴 도메인을, narrow peak 모드에서는 전사인자와 같은 정확한 결합 부위를 각각 최적화된 알고리즘으로 검출합니다. 이러한 다층적 통계 검증 시스템과 실험 유형별 맞춤 모델링을 통해 MACS2는 위양성률을 최소화하면서도 생물학적으로 의미 있는 저농도 결합 부위까지 놓치지 않는 균형잡힌 성능을 제공하여, 후속 주석 분석과 기능적 해석의 신뢰성을 보장하는 ChIP-seq 분석의 필수 핵심 도구로 자리매김하고 있습니다. 입력(input) 데이터로는 Bowtie2를 완료한 SAM 파일을 받고, 출력(output) 데이터로는 peaks.narrowPeak 파일, peaks.xls 파일, summits.bed 파일, model.R 파일이 생성됩니다.
주요사항
- MACS2 callpeak 실행 시 Input (컨트롤) 파일은 파일명이 반드시 CONTROL_로 시작해야 합니다. 반면, Treatment (ChIP) 파일은 별도의 파일명 규칙이 없습니다.
- > Input DNA (컨트롤) 역할 - 비특이적 항체 결합 제거 - 크로마틴 접근성 편향 보정 - PCR 증폭 편향 정규화
실행 명령어 예시
# PEAK_TYPE: tf # (recommended settings for transcription factor ChIP-seq) macs2 callpeak \ -t treatment.bam \ # Treatment file -c control.bam \ # Control file -g hs \ # Genome size -n sample \ # Output prefix --keep-dup 1 \ # Duplicate read handling --broad-cutoff 0.1 \ # Broad peak threshold --broad \ # Broad peak mode -q 0.05 # FDR threshold # PEAK_TYPE: narrow # (recommended settings for narrow peak histone modification ChIP-seq: H3K4me3, H3K27ac) macs2 callpeak \ -t treatment.bam \ -c control.bam \ -g hs \ -n sample \ --nomodel \ # Disable model building --extsize 200 \ # Fragment extension size -q 0.05 # PEAK_TYPE: broad # (recommended settings for broad peak histone modification ChIP-seq: H3K27me3, H3K9me3) macs2 callpeak \ -t treatment.bam \ -c control.bam \ -g hs \ -n sample \ --broad \ # Broad peak mode --broad-cutoff 0.1 \ --nomodel \ --extsize 200 \ -q 0.01 # More stringent FDR
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | ChlP-seq 데이터 디렉토리 경로(sam/bam) | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | String | genome_size | hs | Genome 크기 (-g) (예: 'hs' for human, 'mm' for mouse) | |
| Option | String | peak_type | tf | 피크 타입 최적화 설정 (tf, narrow, broad) |
결과
-
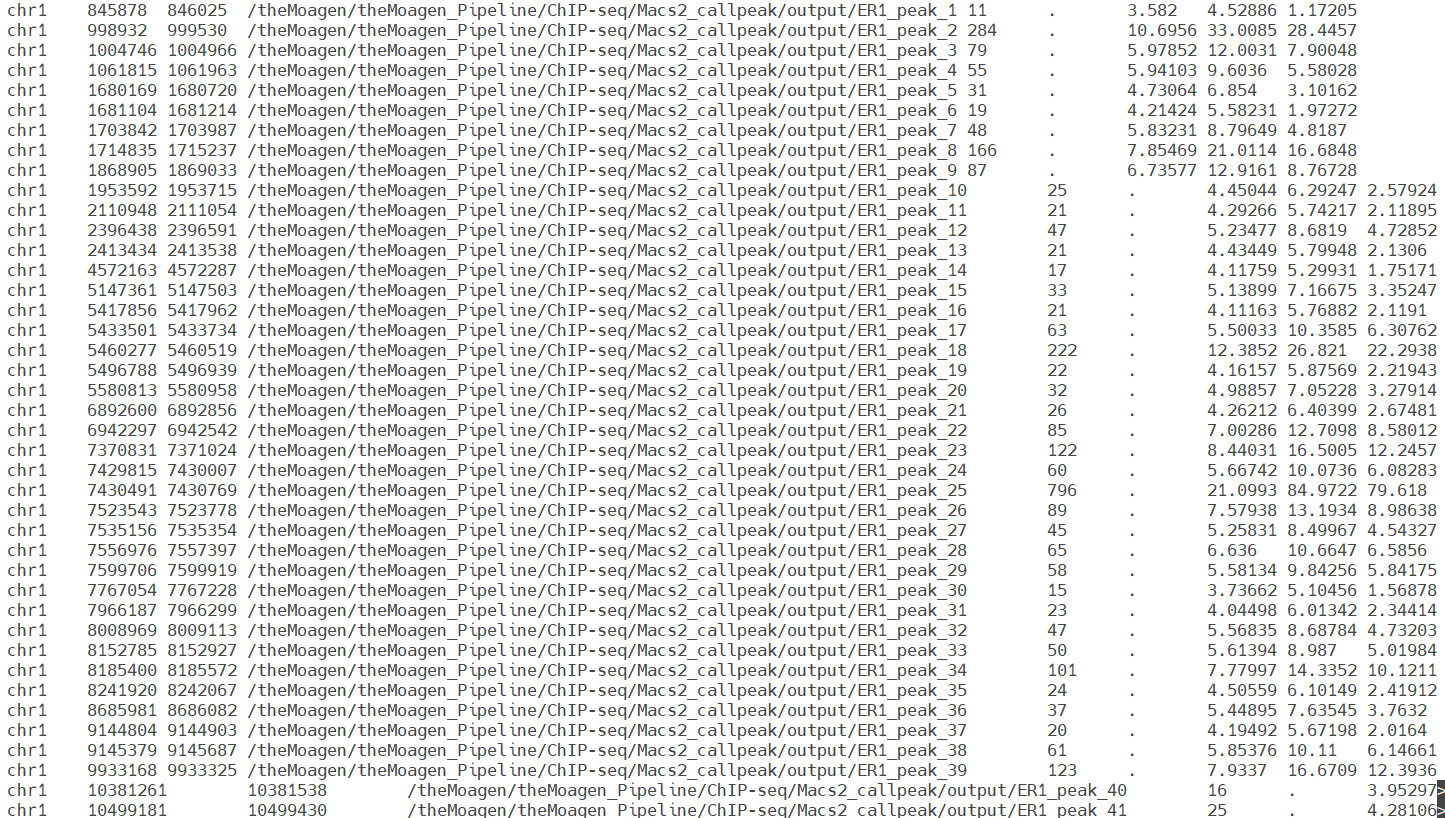
[name]_peaks.broadPeak 파일. 광범위한(broad) peak의 위치를 UCSC Bed 포맷으로 기록한 파일. H3K27me3와 같은 히스톤 마커처럼 넓게 분포하는 신호를 분석할 때 사용됨.
-
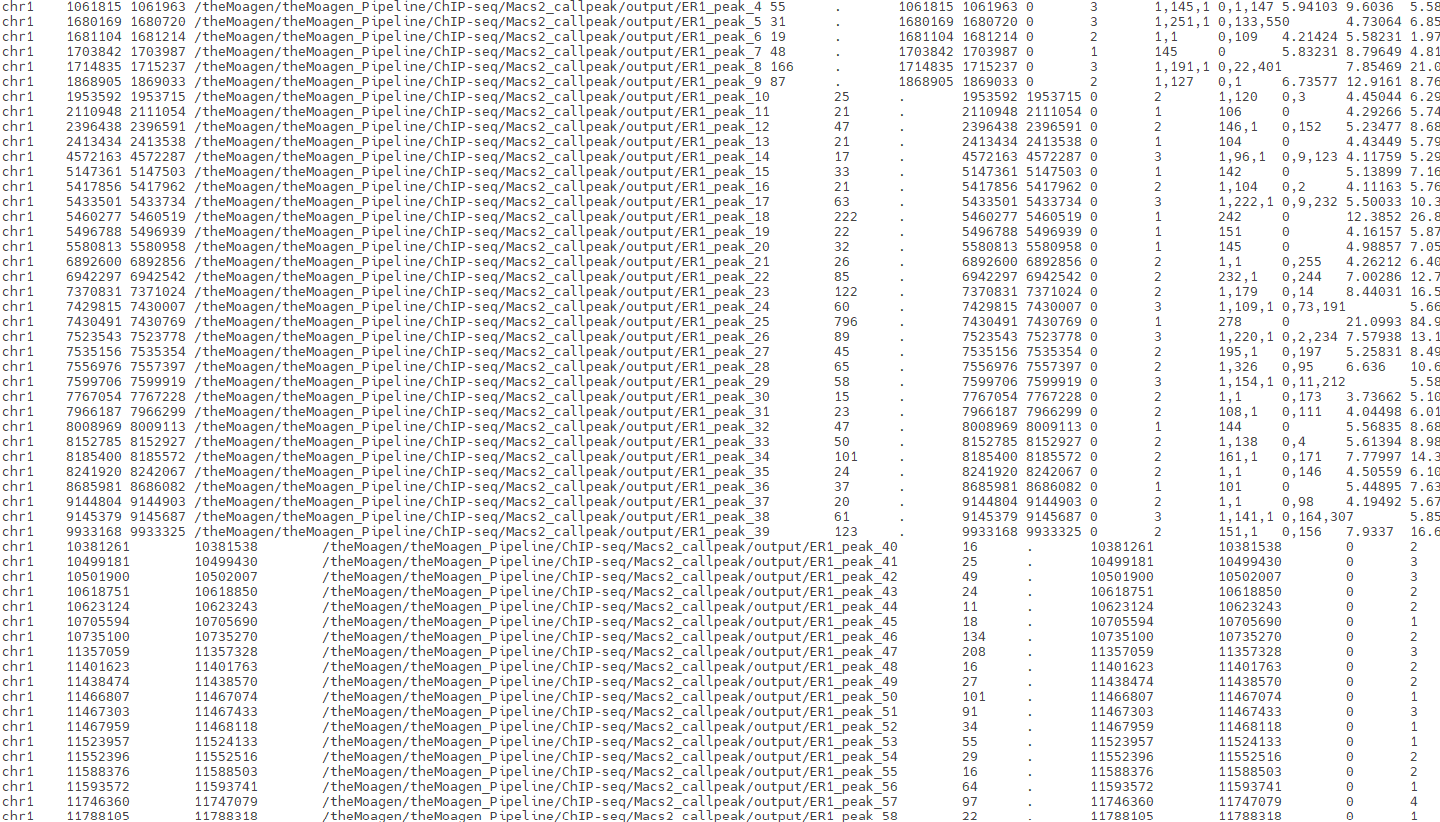
[name]_gappedPeak 파일. 여러 개의 인접한 peak가 합쳐져 하나의 큰 gap이 있는 peak 영역을 형성할 때 사용됨. 개별 피크들의 유전자 위치, 점수, 그리고 피크들이 합쳐진 전체 영역의 정보를 포함함. UCSC Gapped Bed 포맷과 유사함.
-
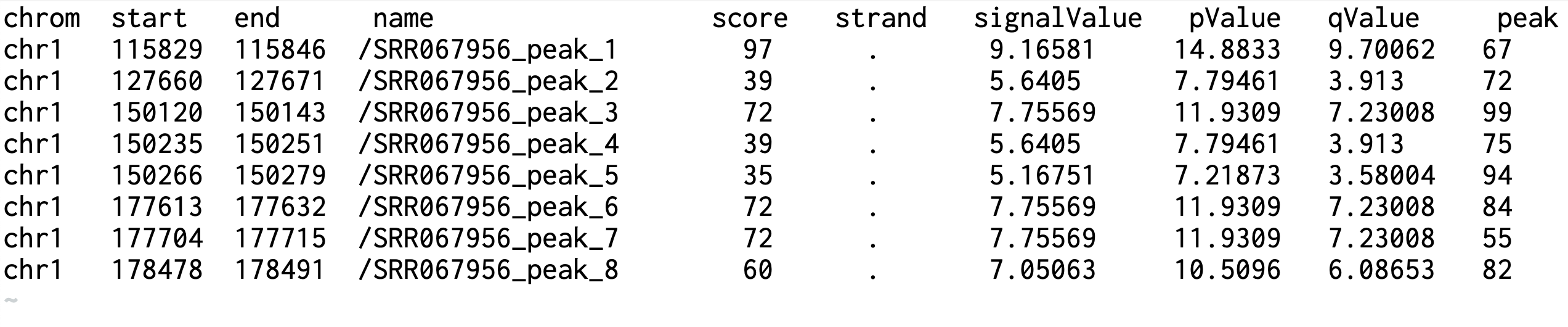
[name]_peaks.narrowPeak 파일. peak 정상부, p-value 및 q-value와 함께 peak의 위치를 포함함. chrom: 염색체 이름을 의미 start: Peak의 시작 위치 end: Peak의 끝 위치 name: Peak에 대한 고유한 이름이나 식별자 score: Peak의 점수 또는 품질 strand: Peak이 위치한 DNA 서열의 방향을 의미 signalValue: Peak에 대한 신호의 값 또는 크기를 의미 pValue: Peak의 p-value를 의미 qValue: Peak의 q-value를 의미 peak: Peak의 위치를 나타냄
-
.png)
[name]_peaks.xls 파일(Header): callpeaks에 대한 정보를 담고 있는 테이블 형식의 파일 Command line: callpeak –t 분석에 사용된 명령어를 보여줌 ARGUMENT LIST: 명령어의 인수들을 설명 name: 분석 결과 파일의 이름을 나타냄 format = AUTO: 파일 형식을 자동으로 인식하도록 지정 Chlp-seq file: Chlp-seq 파일의 경로를 제공 control file: 컨트롤 파일의 경로를 제공 effective genome size: 유효 게놈 크기를 나타냄 band width = 300: Bandwidth를 나타냄 model fold = [5,50]: 모델 폴드의 범위를 나타냄 qvalue cutoff: q-value의 임계값을 나타냄 The maximum gap ...: 유의한 지점간의 최대 간격은 리드 길이 또는 태그 길이로 할당 The minimum length..: Peak의 최소 길이는 예측된 조각 길이 ‘d’로 할당 Larger dataset will ..: 더 큰 dataset은 작은 dataset으로 크기가 조절됨 Range for calculating..: 지역 람다를 계산하는 범위는 1000 bps 와 10000 bps Broad region calling..: 비활성화시 broad peak이 감지되지 않도록 설정 Paired-End mode is off: Paired-End moder가 비활성화 tag size is determined as.: 태그 크기는 36 bps 로 설정 total tags in treatment: 처리 그룹의 전체 태그 수를 나타냄 tags after filtering in ..: 처리 그룹에서 필터링 후의 태그 수를 의미 maximum duplicate tags..: 동일한 위치에서 최대 중복 태그 수를 나타냄 Redundant rate in treat..: 처리 그룹에서의 중복 비율을 나타냄 total tags in control..: control group의 전체 태그 수를 나타냄 tags after filtering in ...: control group에서 필터링 후의 태그 수
-
.png)
_peaks.xls 파일(Main): Main은 call peaks에 대한 정보로 이루어져 있음 chr: 염색체 이름 start: Peak의 시작 위치 end: Peak의 끝 위치 length: Peak의 길이를 나타냄 abs_summit: Peak의 절대적인 부분의 위치 pileup: peak의 높은 밀도 값 fold_enrichment: peak의 fold enrichment 값을 의미
-
.png)
[name]_summits.bed: BED format file indicating the summit position of each peak. chr1: Chromosome information 1158362: Start sequence position of the corresponding region 1158363: End sequence position of the corresponding region /bioex/pipeline..: Peak ID 9.70062: (-Log10)Pvalue
-
.png)
MACS2의 Genome 크기 매개변수
-
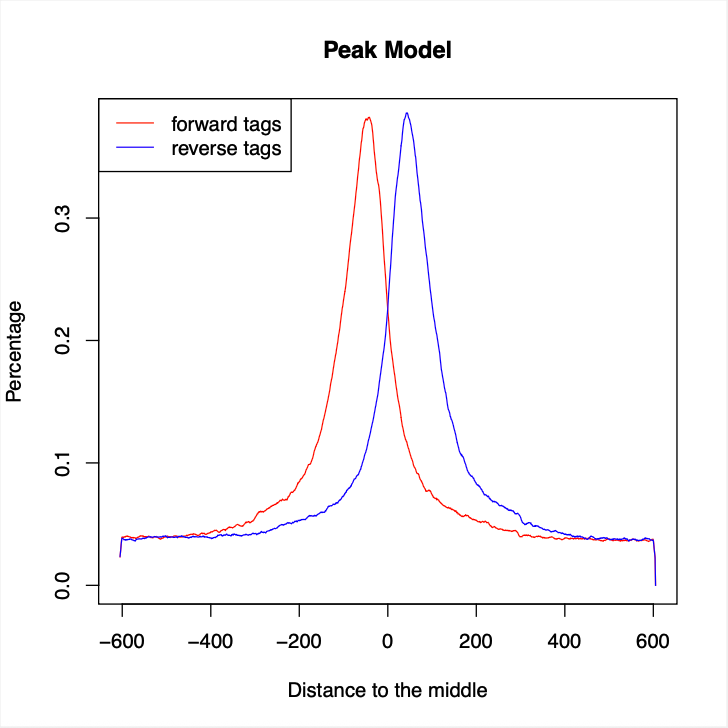
model.R 실행 결과 파일: 데이터에 기반한 모델 및 교차 상관 플롯을 생성하기 위해 사용합니다.
Homer_annotatePeaks
Homer annotatePeaks는 University of California San Diego(UCSD)의 Christopher Benner가 개발한 포괄적 게놈 주석 분석 플랫폼의 핵심 모듈로, 전 세계 ChIP-seq 연구 커뮤니티에서 피크 기능 주석 분야의 표준 도구로 인정받고 있는 강력한 생물학적 해석 엔진입니다. 이 도구는 단순한 거리 기반 주석을 넘어서 계층적 게놈 기능 분류 체계와 다차원 주석 통합 알고리즘을 구현하여, 식별된 ChIP-seq 피크들을 생물학적으로 의미 있는 기능적 카테고리로 체계적으로 분류하고 각 피크의 조절적 역할과 타겟 유전자와의 관계를 정밀하게 규명합니다. Homer annotatePeaks의 가장 큰 혁신은 다중 스케일 주석 시스템(multi-scale annotation system)에 있습니다. 이 도구는 전사 시작점(TSS), 전사 종료점(TTS), 엑손-인트론 경계, 5'/3' UTR 등의 세밀한 유전자 구조 요소들을 동시에 고려하여 각 피크의 위치를 프로모터, 인핸서, 사일렌서, 인슐레이터 등의 기능적 범주로 분류합니다. 특히 거리 가중 주석 알고리즘을 통해 가장 가까운 유전자뿐만 아니라 기능적으로 연관될 가능성이 높은 원거리 유전자까지 포함하는 포괄적인 타겟 유전자 예측을 수행하며, 조직 특이적 유전자 발현 데이터와의 통합을 통해 생물학적 맥락을 고려한 정교한 기능 추론을 제공합니다. Homer annotatePeaks는 GENCODE, RefSeq, Ensembl 등 다양한 유전자 주석 데이터베이스와의 완벽한 호환성을 제공하며, CpG 섬, 반복 서열, 보존된 비코딩 요소, 알려진 전사인자 결합 모티브 등의 다층적 게놈 특성 정보를 통합하여 각 피크의 조절적 잠재력과 진화적 보존성을 종합적으로 평가합니다. 이러한 통합적 주석 접근법을 통해 연구자들은 ChIP-seq 실험에서 발견된 단백질 결합 부위들의 생물학적 의미와 기능적 중요성을 체계적으로 이해하고, 유전자 조절 네트워크와 질병 연관성에 대한 심층적인 인사이트를 얻을 수 있는 필수불가결한 해석 도구입니다. 입력(input) 데이터로는 MACS2가 완료된 summits.bed 파일로 하며, 주석이 달린 peaks의 정보를 포함한 txt file을 출력(output) 데이터로 합니다.
주요사항
- > 주요 게놈 어셈블리 인간 (Homo sapiens) - hg38: GRCh38 (권장, 최신) - hg19: GRCh37 (레거시, 여전히 널리 사용) - hg18: NCBI36 (구형, 특수 목적) 마우스 (Mus musculus) - mm39: GRCm39 (최신) - mm10: GRCm38 (표준) - mm9: NCBI37 (구형) 기타 모델 생물 - dm6: Drosophila melanogaster (초파리) - ce11: Caenorhabditis elegans (예쁜꼬마선충) - danRer11: Danio rerio (제브라피쉬) - rn6: Rattus norvegicus (흰쥐)
실행 명령어 예시
/opt/apps/HOMER/annotatePeaks.pl [peak file].txt hg38 > /path/to/output_dir.tsv
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | /path/to/input_dir | BED 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | /path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | String | genome_size | hg38 | Genome 정보 (hg38, hg19, mm10, etc.) |
결과
-
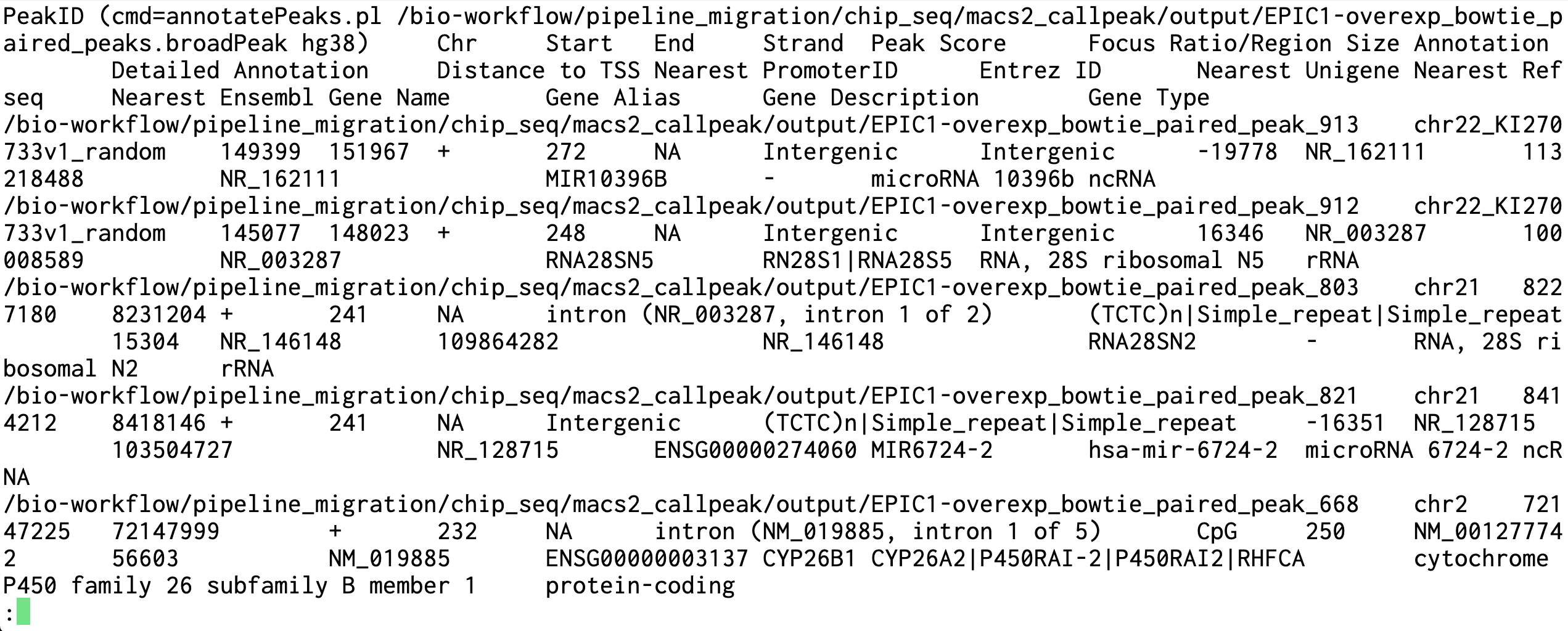
[name]_peaks.broadPeak.txt / [name]_peaks.gappedPeak.txt Peak ID: Chlp-seq 분석에서 각 피크에 대한 고유한 식별자 또는 이름 Chr: 피크가 발견된 염색체 Start: 피크의 시작 위치 End: 피크의 종료 위치 Strand: 피크의 방향 (+ 또는 -) Peak Score: 피크의 점수 또는 통계적인 측정 값 Peak Focus Ratio: 피크의 중심에 대한 양쪽의 비율 Annotation: 피크가 속한 영역의 주석(예: Exon, Intron 등) Detailed Annotation: 상세한 주석 정보(CpG Islands, repeats 등) Distance to TSS: 피크에서 가장 가까운 RefSeq 유전자의 시작 지점까지의 거리 Nearest Promoter ID: 해당 유전자에 대한 가장 가까운 프로모터의 식별자 Entrez ID: Entrez Gene ID로 표시된 유전자의 고유한 식별자 Nearest Unigene: 해당 유전자에 대한 가장 가까운 Unigene ID Nearest Refseq: 해당 유전자에 대한 가장 가까운 RefSeq ID Nearest Ensembl: 해당 유전자에 대한 가장 가까운 Ensembl ID Gene Name: 유전자의 고유한 이름 또는 symbol Gene Alias: 유전자의 다른 이름 또는 별칭 Gene Description: 유전자에 대한 설명 또는 주석 Gene Type: 유전자의 유형 또는 종류(예:protein-coding gene 등)
ROSE
ROSE (Rank Ordering of Super-Ehancer) 는 Whitehead Institute for Biomedical Research의 Young Lab에서 개발한 슈퍼 인핸서 (Super-Enhancer, SEs) 분석 도구입니다. 2013년 Cell 지에 발표된 2개의 슈퍼 인핸서에 대한 논문을 기반으로 하고 있으며, 현재도 전사 조절 및 질병 연구에서 널리 사용되고 있습니다.
인핸서는 DNA 상에 위치하는 50~1500 bp 크기의 짧은 유전자 조절 영역으로, 전사 인자(Transcription Factor) 및 mRNA 전사에 참여하는 여러 단백질 복합체와 상호작용해 타겟 유전자의 전사가 일어날 가능성을 높입니다. 이들은 타겟 유전자와 멀리 떨어진 곳에 위치하지만, 단백질들에 의해 DNA가 3차원 loop 구조를 이루면 이들에 직접 작용하며 전사 작용에까지 영향을 미칩니다. 수많은 인핸서들이 게놈에 존재하며 세포 및 조직 특이적인 유전자 발현 패턴을 결정하는 데 중요한 역할을 합니다.
슈퍼 인핸서는 그 중에서도 작용이 강한 영역입니다. 보통 여러 인핸서가 모여 있어 상대적으로 긴 영역을 가집니다. 또한 세포의 특정 기능을 수행하는 핵심 유전자들의 발현을 조절하는 인핸서들로 구성되어 있어 흔히 세포의 정체성(Cell Identity)을 결정한다고 표현합니다. 때문에 암과 같은 질병 연구에도 활발히 활용되고 있습니다.
ROSE는 ChIP-seq의 시그널 밀도를 분석하고 순위를 매기는 기법을 사용하여, 유전자 발현을 강력하게 조절하는 슈퍼 인핸서를 다른 일반 인핸서들로부터 구분 합니다. 구체적인 방식은 다음과 같습니다. 먼저 가까이 있는 작은 인핸서 후보 영역들을 하나로 꿰매어(stitch) 더 큰 영역을 만듭니다. 그다음, 이 꿰맨 영역들 각각에 결합된 단백질의 양(ChIP-seq 신호)을 측정하고 순위를 매깁니다 (Rank Ordering). 이 순위별 신호량을 그래프로 그렸을 때, 신호량이 급격히 증가하는 변곡점을 기준으로 슈퍼 인핸서로 분류합니다.
ROSE는 크게 2가지 유형의 입력 파일을 받습니다. 첫 번째는 시그널의 매핑 정도를 볼 수 있는 BAM 파일입니다. 이 파일은 인핸서의 활성도를 정량화하는 데 사용되며, 분석 전에 samtools와 같은 도구를 이용해 반드시 Sorting 및 Indexing 처리가 이루어져야 합니다. 두 번째는 시그널 calling을 통해 선정된 영역 정보가 있는 GFF 파일입니다. 이 파일은 ROSE가 인핸서의 구성 요소로 사용할 게놈 상 위치를 제공하며, 이번 파이프라인의 경우 MACS2 callpeak 출력 결과 중 .broadPeak 파일을 사용했습니다. 이는 분석 이후에는 슈퍼 인핸서의 위치 정보와 정량적 정보를 담은 BED, 혹은 텍스트 파일로 출력됩니다.
실행 명령어 예시
./ROSE.sh \ input_bam="./bowtie2/output" \ input_peak="./macs2_callpeak/output/EPIC1-overexp_bowtie_paired_peaks.broadPeak" \ output_dir="./rose/output/" \ genome="HG38"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | bowtie2/output | bam 및 bai 파일이 있는 디렉토리 경로. bam 파일은 samtools와 같은 도구로 정렬되어 있어야 하며, sorted.bam으로 끝나는 파일명을 가짐. 이에 대한 indexing 파일인 sorted.bam.bai 파일도 요구됨. | |
| Input | File | input_peak | macs2_callpeak/output/EPIC1-overexp_bowtie_paired_peaks.broadPeak | macs2_callpeak의 결과물인 .broadPeak 혹은 .narrowPeak 파일의 경로 | |
| Output | Folder | output_dir | rose/output | 실행 후 결과물을 저장할 디렉토리 경로 | |
| Option | String | genome | HG38 | 사용한 ucsc 게놈의 버전(MM9,MM8,HG18,HG19,HG38) |
결과
-
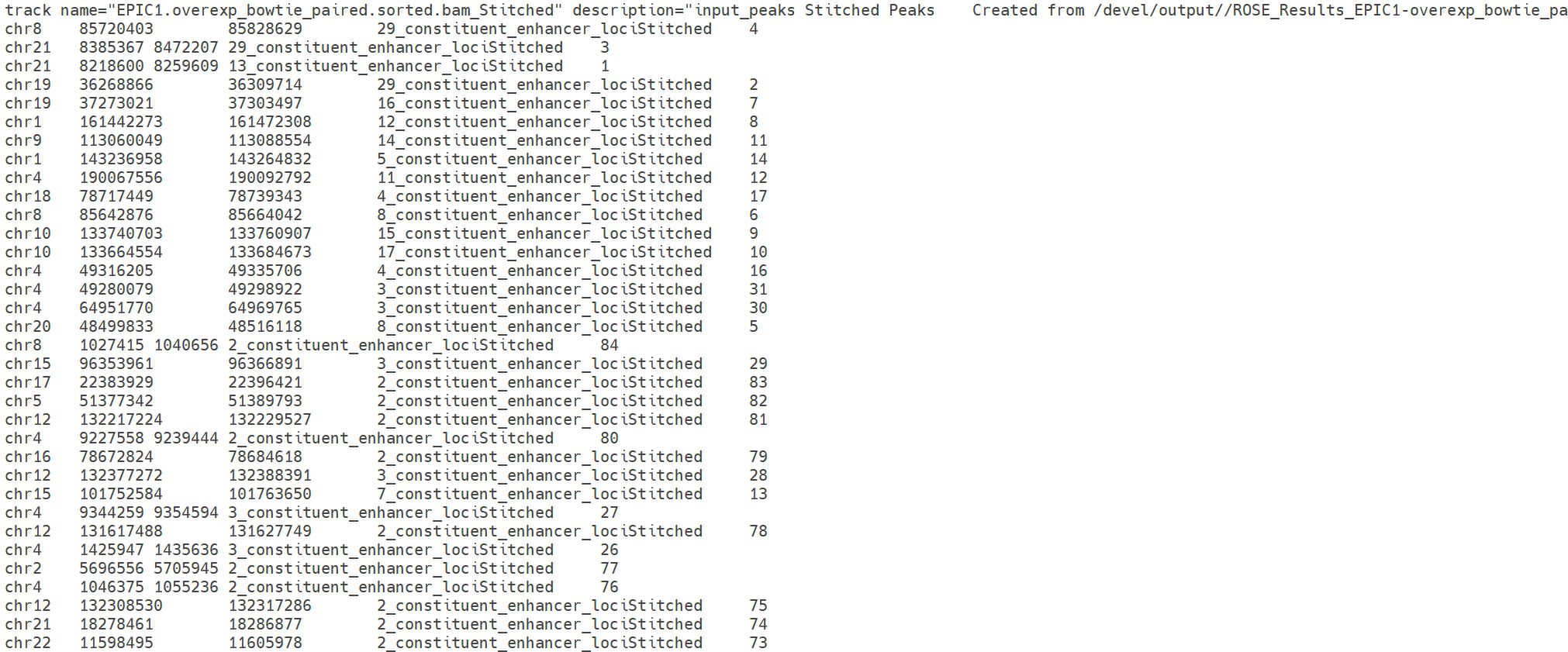
Stitched_withSuper.bed. super-enhancer 영역 정보가 담긴 bed 파일
-
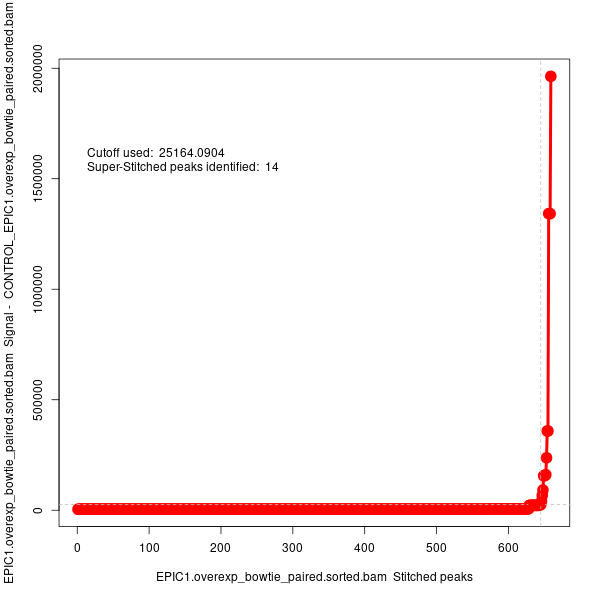
peaks_Plot_points.png. 각 peak의 강도를 시각화한 plot으로, super-enhancer를 선별한 cutoff 기준을 확인할 수 있음.
-
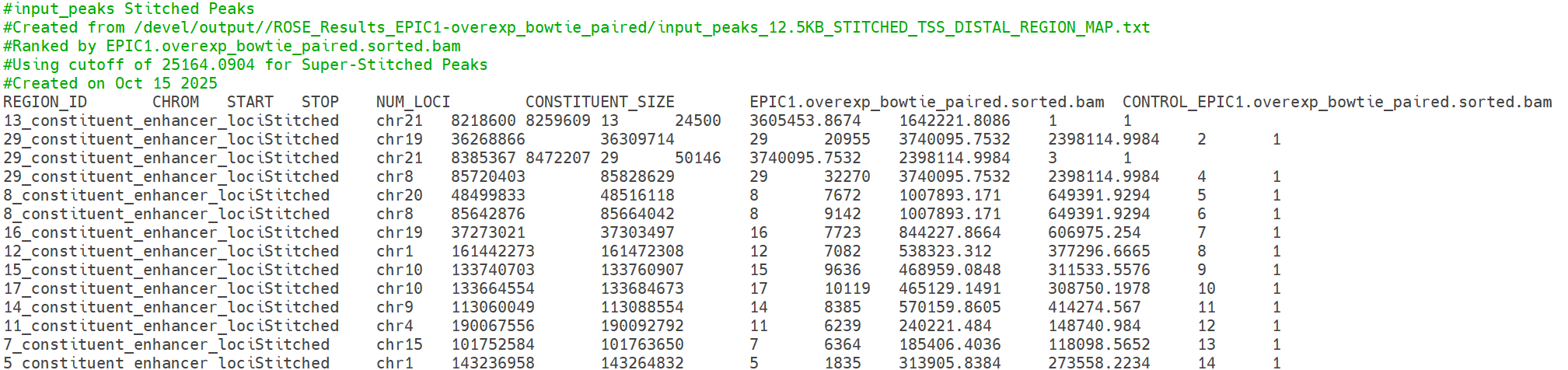
SuperStitched.table. super-enhancer 영역의 좌표, 크기, signal, 밀도 등의 정보가 정리된 표.
-
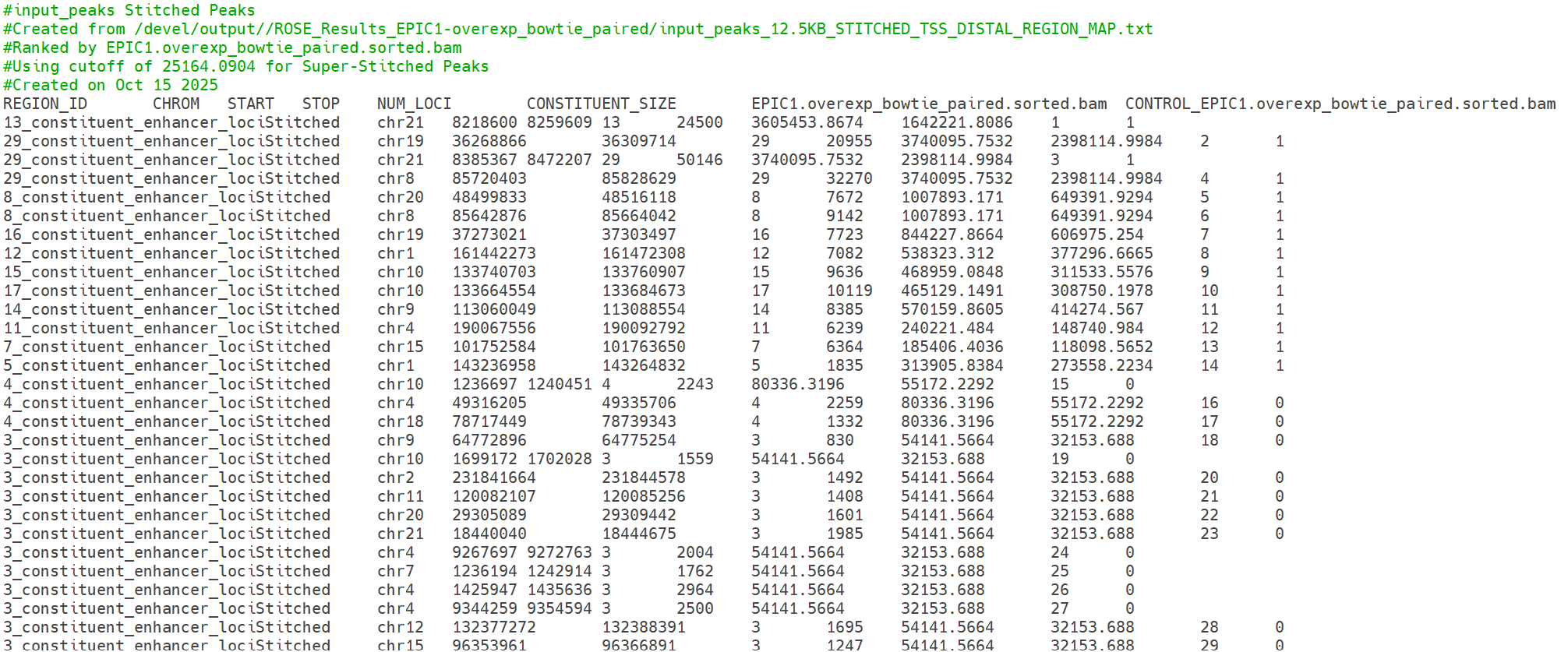
AllStitched.table. super-enhancer 및 모든 stitched enhancer 영역에 대한 정보가 정리된 표.
-
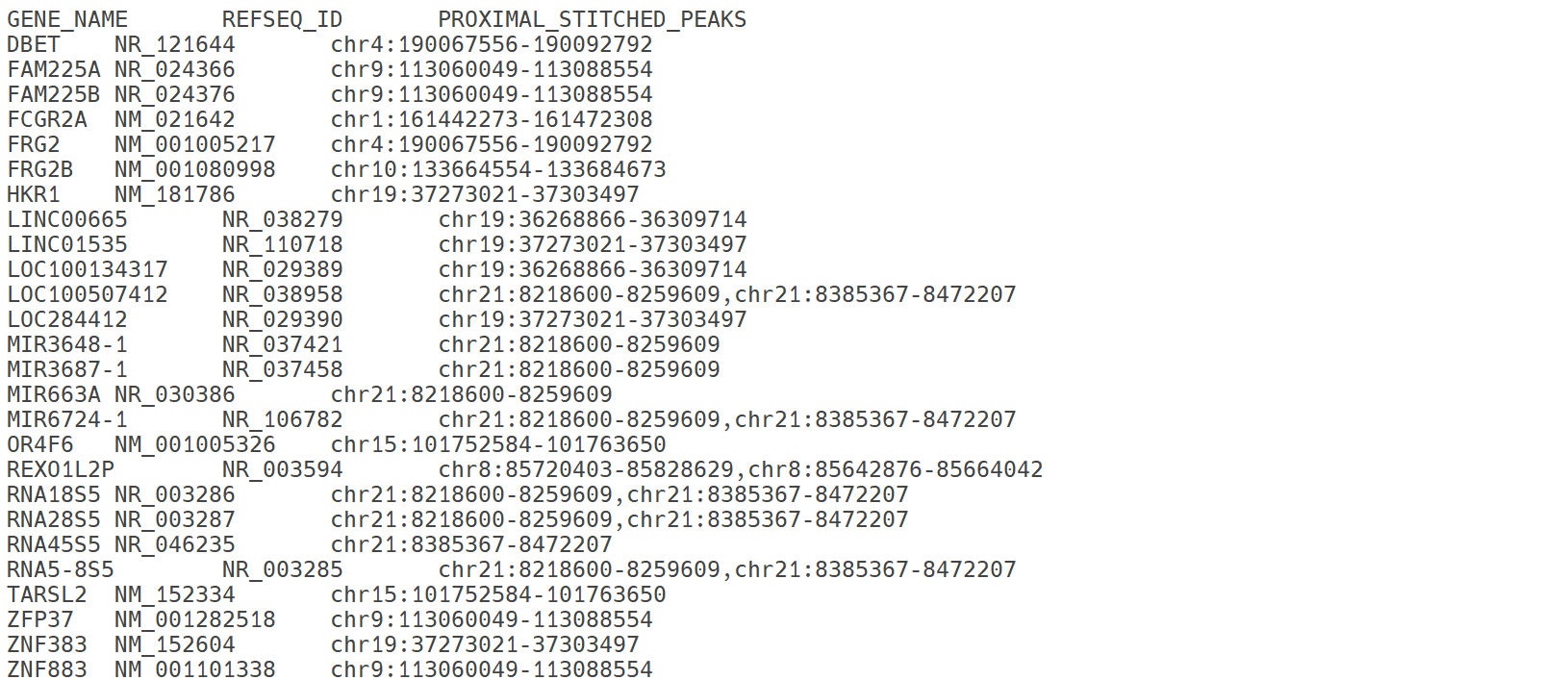
SuperStitched_GENE_TO_REGION. 각 super-enhancer에 연결된 것으로 예측되는 유전자 목록을 보여줌.
-
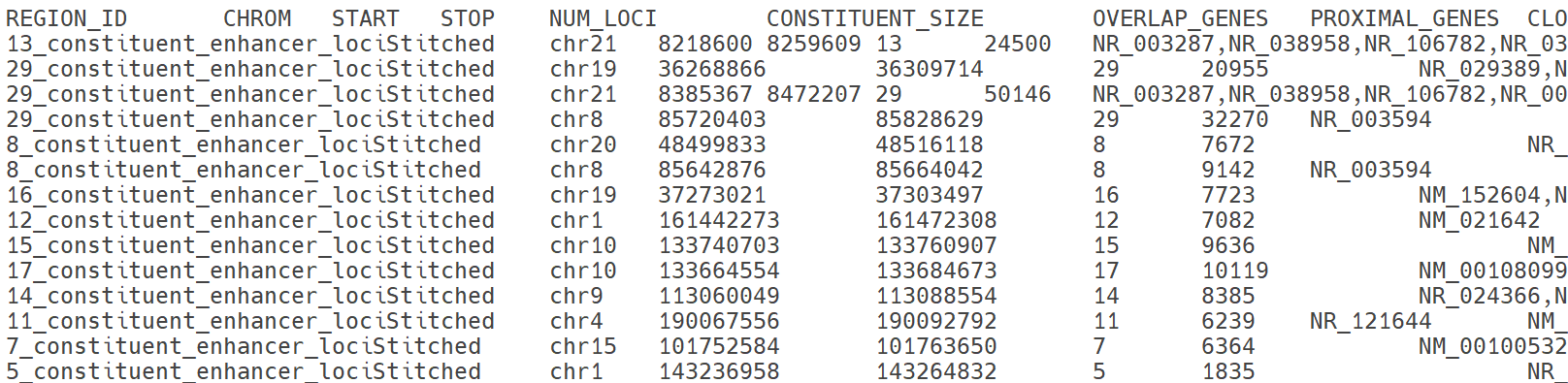
SuperStitched_REGION_TO_GENE. super-enhancer 영역 목록과 각 영역이 연결된 유전자를 쌍으로 보여주는 매핑 파일.
-
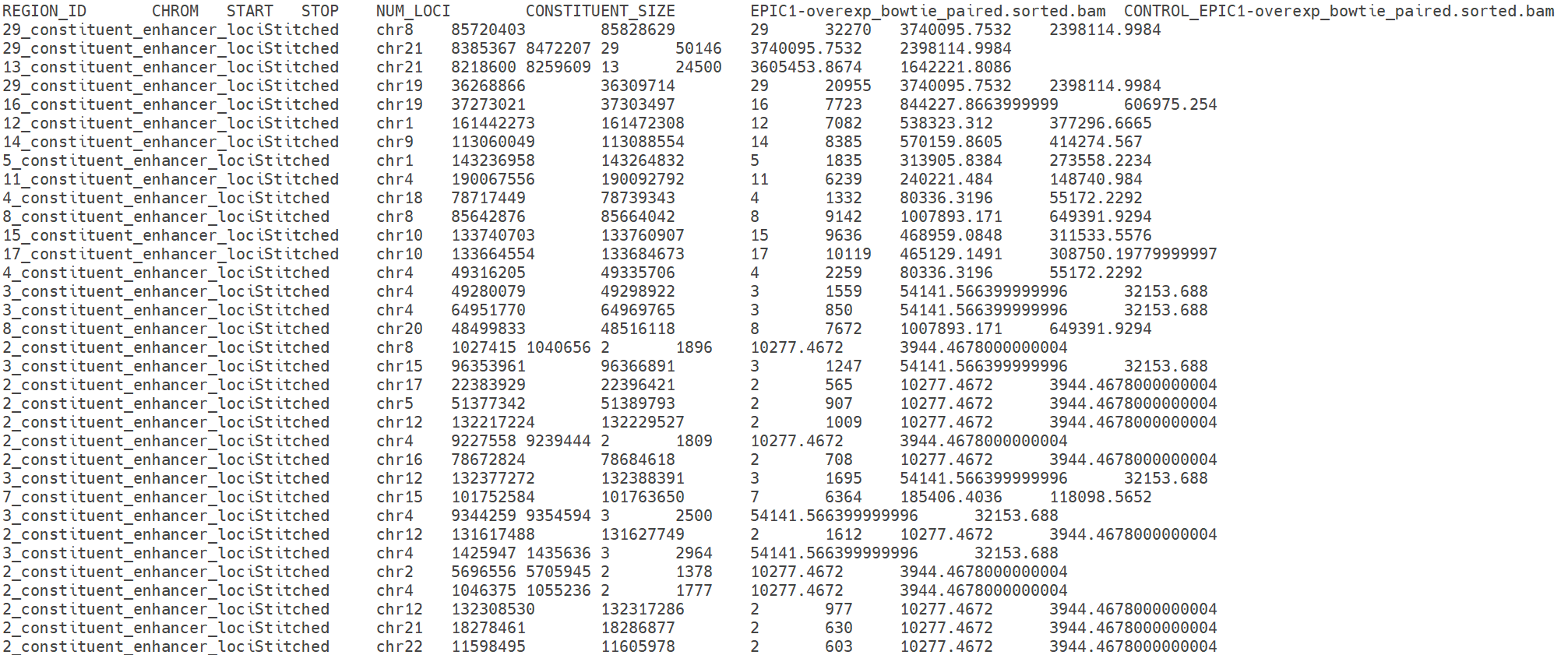
STITCHED_TSS_DISTAL_REGION_MAP. TSS (Transcription Start Site)에서 멀리 떨어진 Stitched Enhancer 영역들을 매핑하여 보여줌.
- 버전1.0
- 마지막 업데이트1일 전
- 기여자
- 이
- 더
- 김
- 클
- 이
- 유