- 카테고리 Transcriptomics > Single-cell-transcriptomics
- 수정일2025-09-23 15:22:34
- 레퍼런스
Single-cell RNA sequencing pipeline은 10X Genomics의 Cell Ranger, python 패키지인 Scanpy를 이용하여 단일 세포 수준의 유전자 발현 데이터를 분석하기 위한 파이프라인입니다. 본 파이프라인은 raw sequencing data(FASTQ 파일)과 함께 각 library가 유래한 샘플의 정보를 함께 입력받습니다. 이후 count matrix 생성, data preprocessing, normalize and scaling, dimension reduction and clustering, annotation, differential gene expression analysis 및 functional analysis 등을 수행합니다. 결과물로 cell-by-gene expression matrix 및 이로부터 파생된 분석 결과 테이블, 시각화한 그래프 등을 얻을 수 있습니다.
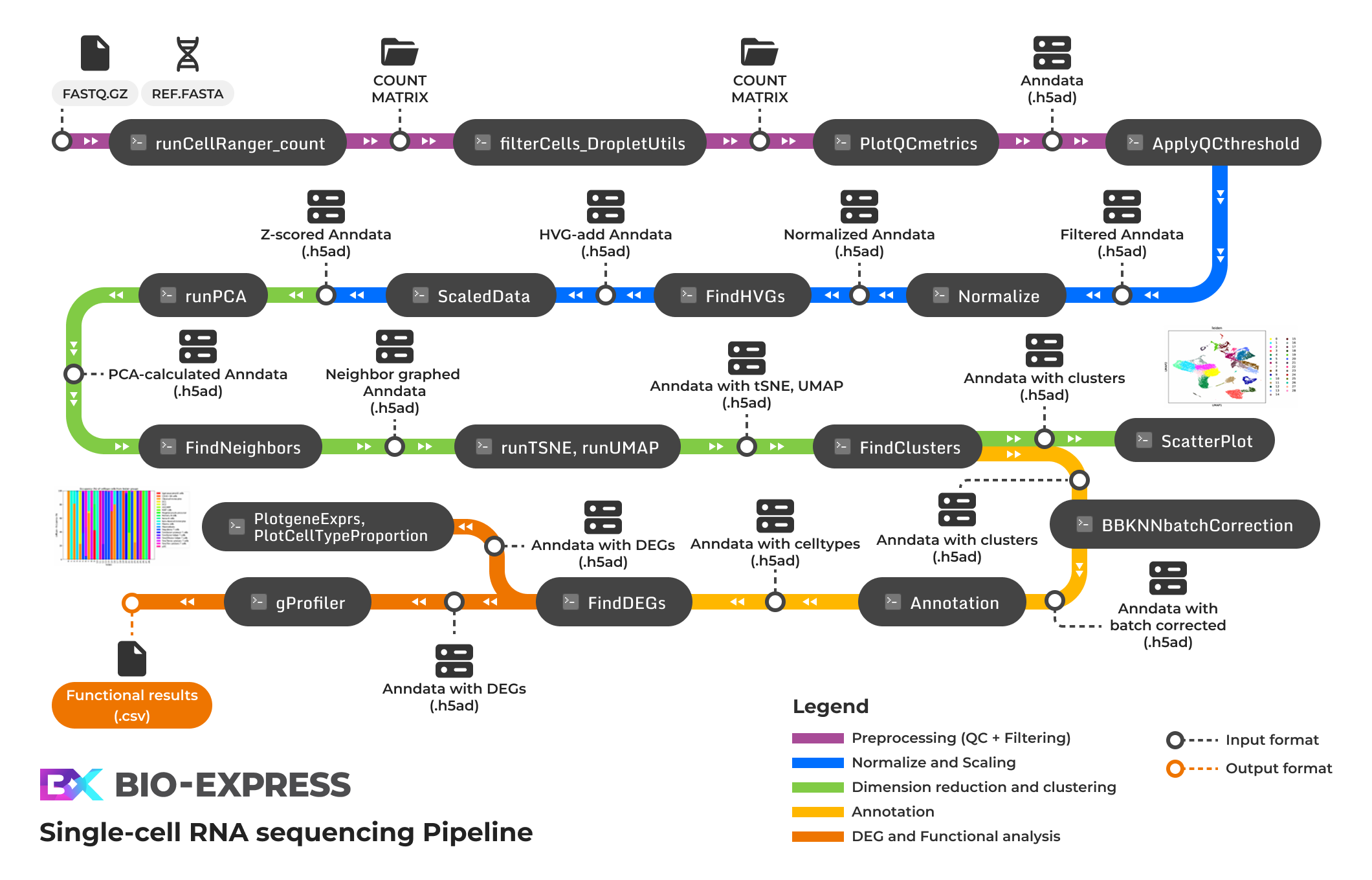
파이프라인 모듈
cellranger_mkref
Cell Ranger는 10x Genomics 사의 single-cell library preparation kit로 제작된 scRNA-seq 라이브러리에서 생성된 FASTQ 파일을 처리하는 소프트웨어입니다. 후술할 cellranger count 단계에서는 read alignment를 수행하고 feature-by-barcode 기반의 UMI(unique molecular identifier) count matrix를 생성합니다. 이를 위해서는 reference genome (FASTA)과 gene annotation (GTF) 파일이 필요하며, 이 파일들을 Cell Ranger가 읽을 수 있도록 전용 형식으로 미리 변환되어야 합니다. 이 작업은 cellranger mkref 단계에서 수행됩니다.Bio-Express에는 GRCh38 (Homo sapiens) version 44와 GRCm39 (Mus musculus) version M33에 대한 reference가 이미 내장되어 있으므로, 해당 reference를 사용할 경우 runCellRanger_mkref 단계를 생략할 수 있습니다.
주요사항
- 10x Genomics에서 제공하는 공식 reference (GRCh38, mm10 등)는 인간과 마우스에 한정되기 때문에 cellranger mkref를 사용하면 임의의 유전체 + GTF 파일을 기반으로 커스텀 reference를 생성할 수 있어, 비모델 생물(non-model species), 박테리아, 곰팡이, 어류 등에도 적용 가능합니다.
입력 데이터 예제
실행 명령어 예시
python cellranger mkref.py genome = “custom” \ fasta = ./genome.fa \ genes = ./genes.gtf output_dir = ./cellranger_mkref/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | String | genome | custom | 결과 파일들이 담길 폴더의 이름 (알파벳, 숫자, “_”, “-”로만 조합하여 작성) | |
| Input | File | fasta | ./genome.fa | FASTA 파일의 경로 | |
| Input | File | genes | ./genes.gtf | GTF 파일의 경로 | |
| Output | Folder | output_dir | ./cellranger_mkref/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | ref_version | v1 | Reference 내에 저장될 reference 버전 정보. |
결과
-
cellranger mkref 결과 폴더 구조
-

reference.json 파일 내용. 이 파일은 mkref 실행에 관련된 메타데이터 정보를 보여 줌
cellranger_count
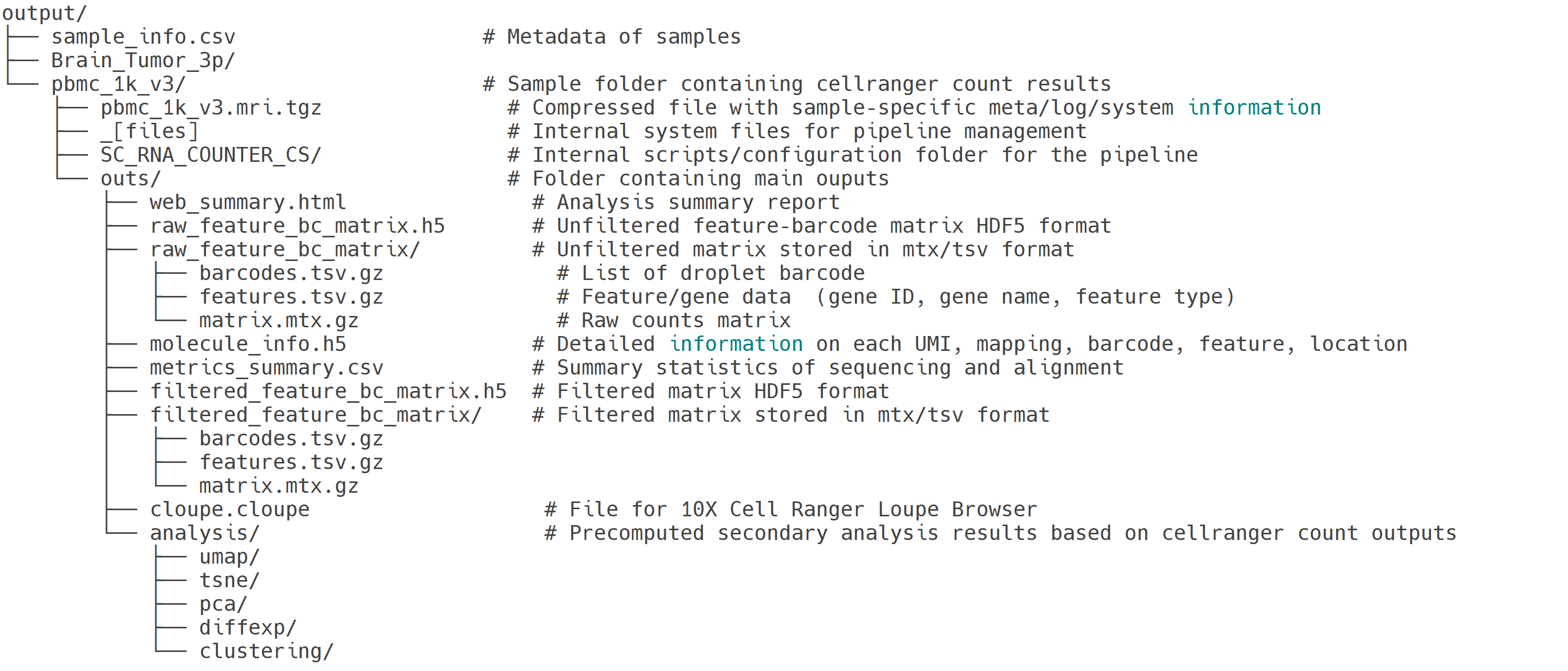
Cellranger count 단계는 Cell Ranger의 count 서브커맨드를 이용하여 단일 scRNA 샘플(single sample)에 대해 read alignment, barcode 및 UMI 처리, 유전자 발현 정량화, 품질 지표 생성 등 전체 분석 과정을 자동으로 수행합니다. 이 과정은 10X Chromium 플랫폼에서 제작된 scRNA-seq 라이브러리로부터 생성된 FASTQ 파일을 입력으로 받아, gene-by-cell expression matrix를 산출합니다. 또한 각 샘플의 메타데이터를 표 형식으로 저장하여 후속 분석에 활용할 수 있습니다.
주요사항
- cellranger count는 --id=output_dir_name 옵션으로 출력 디렉토리 이름만 지정할 수 있으며, 경로 지정은 지원하지 않습니다. 따라서 실행 결과는 output_dir/id 위치에서 확인이 가능합니다.
입력 데이터 예제
실행 명령어 예시
python cellranger_count.py id = pbmc_1k_v3,Brain_Tumor_3p sample = pbmc_1k_v3,Brain_Tumor_3p \ fastqs = ./fastq.gz \ transcriptome = ./cellranger_mkref/output \ output_dir = ./cellranger_count/output \ add_condition = condition,group1,group2 \ description = None, None \ project = None \ lanes = None \ libraries = None \ feature_ref = None \ tenx-cloud-token-path = None \ cell-annotation-model = None \ expect-cells = None \ force-cells = None \ r1-length = None \ r2-length = None \ include-introns = FALSE \ chemistry = None, None \ check-library_compatibility = None \ secondary = None \ min_crispr_umi = None \ no_libraries = None
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | String | id | pbmc_1k_v3,Brain_Tumor_3p | (여러 샘플의 경우 공백 없이 쉼표로 구분) 고유한 실행 ID 문자열 (예: sample345). 이 이름은 임의로 지정할 수 있으며, 모든 출력 파일을 포함하는 디렉토리 이름으로 사용됨. 영문자, 숫자, 밑줄(_), 하이픈(-)만 허용되며, 샘플 당 최대 64자까지 가능 | |
| Input | String | sample | pbmc_1k_v3,Brain_Tumor_3p | (여러 샘플의 경우 공백 없이 쉼표로 구분) 선택할 FASTQ 파일의 파일명 접두어 | |
| Input | Folder | fastqs | ./fastq.gz | (여러 샘플의 경우 공백 없이 쉼표로 구분) 입력 FASTQ 데이터의 경로. 전체 경로에는 쉼표를 포함할 수 없음 | |
| Input | String | transcriptome | ./cellranger_mkref/output | cellranger mkref를 통해 생성된 reference 파일이 포함된 디렉토리 경로. 값이 None이면서 species가 human 혹은 mouse인 경우 Bio-Express 내 reference 파일을 사용함 | |
| Input | String | species | human | 생물 종 선택: 'human' 또는 'mouse' [transcriptome]을 지정하지 않은 경우 필수 | |
| Output | Folder | output_dir | ./cellranger_count/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | add_condition | condition,group1,group2 | (공백 없이 쉼표로 구분) 샘플에 대한 사용자 정의 메타데이터. 예를 들어, 두 가지의 메타데이터 정보와 네 개의 샘플에 대하여, 'disease,cancer,cancer,normal,normal,batch,1,2,3,4'를 입력하는 경우 disease 필드와 batch 정보에 cancer/normal 혹은 1~4의 batch 정보를 tabular format으로 생성함 | |
| Option | String | description | None, None | (여러 샘플에 대해 쉼표로 구분된 형식) 출력 파일에 포함할 샘플 설명입니다. 각 설명에는 쉼표가 포함되지 않아야 합니다. | |
| Option | String | project | None | mkfastq 또는 bcl2fastq 생성 폴더 내에서 FASTQ를 선택할 프로젝트 폴더의 이름 | |
| Option | String | lanes | None | (여러 샘플에 대해 쉼표로 구분된 형식) 선택한 차선의 FASTQ만 사용 | |
| Option | String | libraries | None | 입력 라이브러리 데이터 소스를 선언하는 CSV 파일 | |
| Option | String | feature_ref | None | 기능 참조 CSV 파일, 기능 바코드 구조 및 관련 바코드 선언 | |
| Option | String | tenx_cloud_token_path | None | Cell anotation을 활성화하는 데 사용되는 10배 클라우드 분석 사용자 토큰의 경로입니다. 제공되지 않으면 Cellranger 클라우드 인증 설정을 통해 저장된 위치로 기본 설정됩니다. 토큰을 생성하거나 액세스하는 방법을 알아보세요. | |
| Option | String | cell_annotation_model | None | 사용할 Cell annotation 모델. 유효한 모델 이름은 'auto'와 'human_pca_v1_beta'입니다. 지정되지 않은 경우 셀 주석을 실행하지 않습니다. | |
| Option | String | expeforce_cellsct_cells | None | (여러 샘플에 대해 쉼표로 구분된 형식) 셀 호출 알고리즘의 입력으로 사용되는 예상 복구 셀 수 | |
| Option | String | force_cells | None | (여러 샘플에 대해 쉼표로 구분된 형식) 파이프라인이 셀 호출 알고리즘을 우회하여 이 셀 수를 사용하도록 강제합니다. [최소: 10] | |
| Option | String | r1_length | FALSE | (여러 샘플에 대해 쉼표로 구분된 형식) 분석 전에 입력 읽기 1을 이 길이로 하드 트림합니다 | |
| Option | String | r2_length | None,None | (여러 샘플에 대해 쉼표로 구분된 형식) 분석 전에 입력 읽기 2를 이 길이로 하드 트림합니다 | |
| Option | String | include_introns | None | (여러 샘플에 대해 쉼표로 구분된 형식) 분석 전에 입력 읽기 1을 이 길이로 하드 트림합니다 | |
| Option | String | chemistry | None | (여러 샘플에 대해 쉼표로 구분된 형식) 분석 구성. 참고: 기본적으로 분석 구성은 자동으로 감지되며, 이는 권장 모드입니다. 일반적으로 화학 물질을 지정할 필요는 없습니다. 옵션은 다음과 같습니다: 'auto' for autodetection, 'threeprime' for Single Cell 3', 'fiveprime' for Single Cell 5', 'SC3Pv1' or 'SC3Pv2' or 'SC3Pv3' or 'SC3Pv4' for Single Cell 3' v1/v2/v3/v4, 'SC3Pv3LT' for Single Cell 3' v3 LT, 'SC3Pv3HT' for Single Cell 3' v3 HT, 'SC5P-PE' or 'SC5P-PE-v3' or 'SC5P-R2' or 'SC5P-R2-v3', for Single Cell 5', paired-end/R2-only, 'SC-FB' for Single Cell Antibody-only 3' v2 or 5'. 멀티놈 데이터의 GEX 부분을 분석하려면 화학 물질을 'ARC-v1' [기본값: auto]로 설정해야 합니다 | |
| Option | String | check_library_compatibility | None | 플롯 출력 파일의 확장. | |
| Option | String | secondary | None | 클러스터링과 같은 2차 분석을 수행합니다. | |
| Option | String | min_crispr_umi | None | M(여러 샘플에 대해 쉼표로 구분된 형식) 최소 CRISPR UMI 임계값 [기본값: 3] | |
| Option | String | no_libraries | None | --feature-ref를 사용하지만 '라이브러리' 플래그가 지정된 기능 바코드 라이브러리는 사용하지 않습니다 |
결과
-

sample_info.csv 파일 내용. 샘플별 사용자 정의 메타데이터를 포함하는 CSV 파일
-
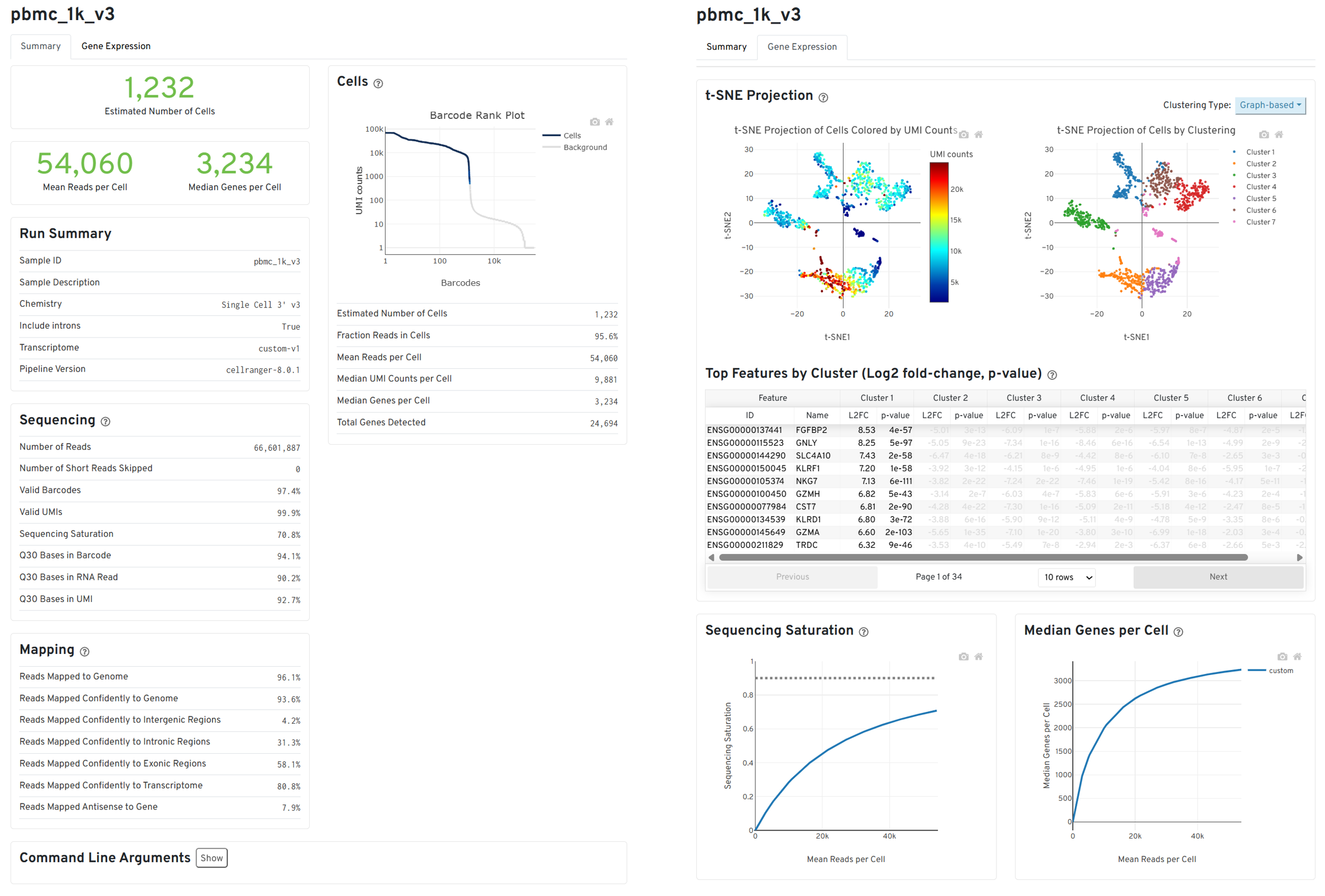
web_summary.html 파일 내용. cellranger count 명령어 결과 요약 리포트로 파일은 분석된 단일 세포 RNA 시퀀싱 데이터에 대한 주요 품질 지표 및 요약 정보를 시각적으로 제공함
-
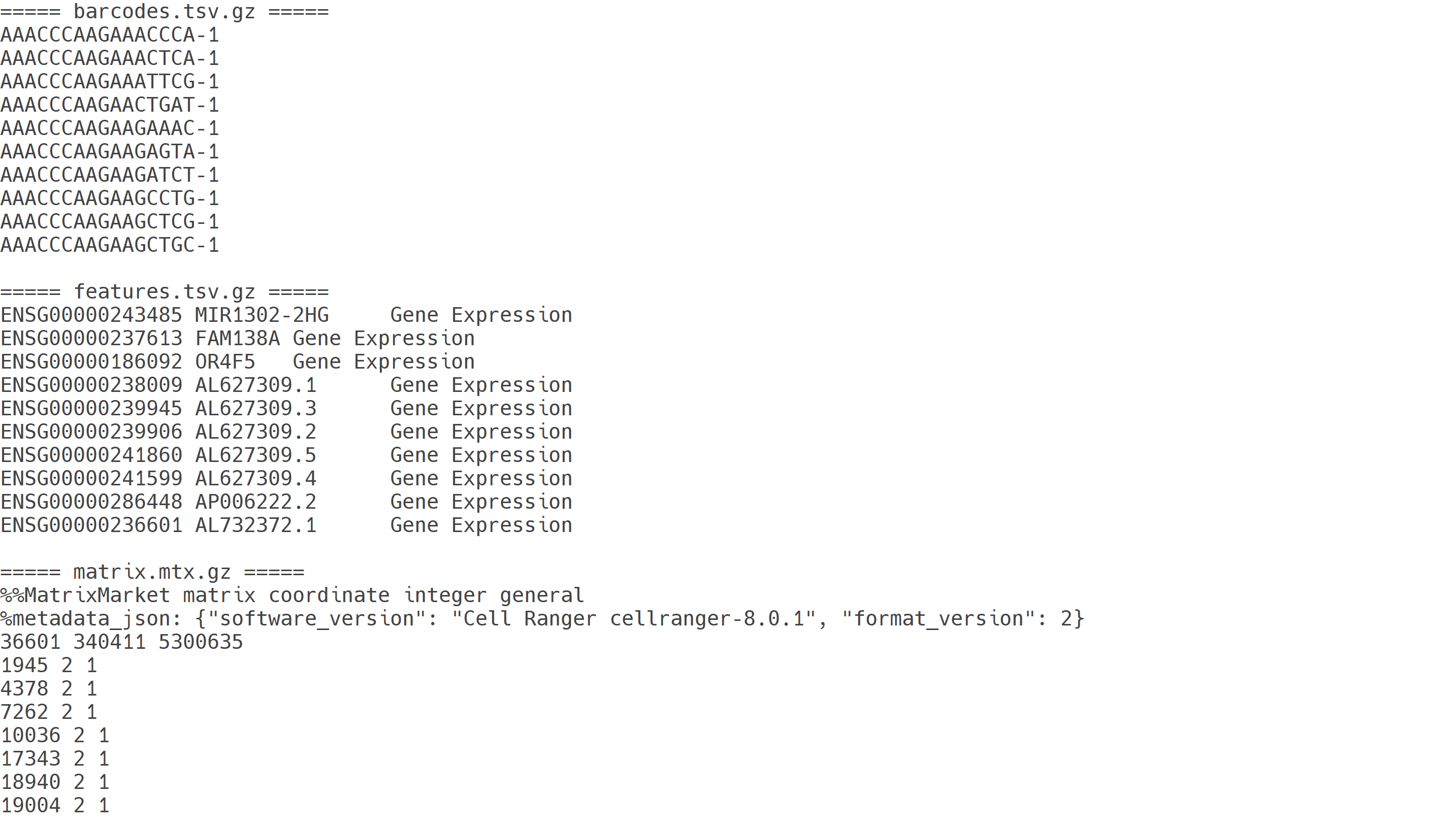
raw_feature_bc_matrix 폴더 내용. 분석용 발현 행렬로 세포 바코드 목록(barcodes.tsv.gz), 유전자(또는 feature) 정보(features.tsv.gz), 세포 x 유전자 발현 데이터(matrix.mtx.gz)로 구성됨
-
.png)
cellranger count 결과 폴더 구조
dropletUtils_filterCells
DropletUtils의 filterCells는 sample_info.csv 파일을 불러온 뒤, 해당 파일에 기재된 각 샘플에 대해 비어 있는 droplet을 제거합니다. 구체적으로는 다음 절차를 거쳐 수행됩니다. 먼저, cellranger count의 산출물 중 filtering되지 않은 feature-barcode count matrix를 불러오고, 이 과정에서 각 droplet에 sample_info.csv에서 얻은 샘플 정보를 매핑하여 저장합니다. 이후 각 droplet의 총 UMI 수를 계산하여 기본값으로 100 이하인 경우를 비어 있는 droplet으로 간주합니다. 이러한 기준을 바탕으로 전체 droplet 중 실제로 empty droplet일 것으로 추정되는 경우를 선별하고, 반대로 비어 있지 않은 droplet은 실제 세포로 판단합니다. 실제 세포로 구분된 droplet의 count matrix는 추후 분석에 활용할 수 있도록 입력과 동일한 .mtx 형식으로 저장합니다. 또한 filtering되지 않은 전체 droplet을 대상으로 barcode rank plot을 작성하여 제거된 droplet의 UMI 분포와 rank 정보를 시각적으로 확인할 수 있습니다.
주요사항
- DropletUtils::read10xCounts() 또는 read10xRaw()는 10X Genomics 형식에 최적화되어 있으므로, 비표준 형식(예: custom scRNA-seq protocol)을 사용하는 경우 별도 전처리가 필요합니다. 또한 입력 데이터 포맷으로 cellranger count의 unfiltered matrix (raw_feature_bc_matrix)를 사용해야 합니다.
입력 데이터 예제
실행 명령어 예시
Rscript DropletUtils_filterCells.r input_dir = ./cellranger_count/output \ output_dir = ./dropletutils_filtercells/output \ lower = 100 \ niters = 10000 \ test_ambient = False \ ignore = NULL \ retain = NULL \ fdr_thr = 0.05
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./cellranger_count/output | 입력 파일이 저장된 디렉토리 경로로 cellranger count 결과 폴더임 | |
| Output | Folder | output_dir | ./dropletutils_filtercells/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Integer | lower | 100 | 총 UMI count의 하한값. 이 값 이하의 barcode는 빈 droplet(empty droplet)으로 간주함 | |
| Option | Integer | niters | 10000 | Monte Carlo p-value 계산을 위한 반복 횟수 | |
| Option | Boolean | test_ambient | False | True인 경우 lower 값 이하의 barcode도 검정에 포함하여 결과를 반환함 | |
| Option | Integer | ignore | NULL | 총 UMI count의 하한을 지정, 이 값 이하인 barcode는 무시됨 | |
| Option | Integer | retain | NULL | 총 UMI count의 이 값을 초과하는 barcode는 세포를 포함한다고 가정 | |
| Option | String | fdr_thr | 0.05 | Monte Carlo test로 산출된 p-value에 기반해 low-quality cell을 필터링할 때 사용하는 FDR 임계값 |
결과
-
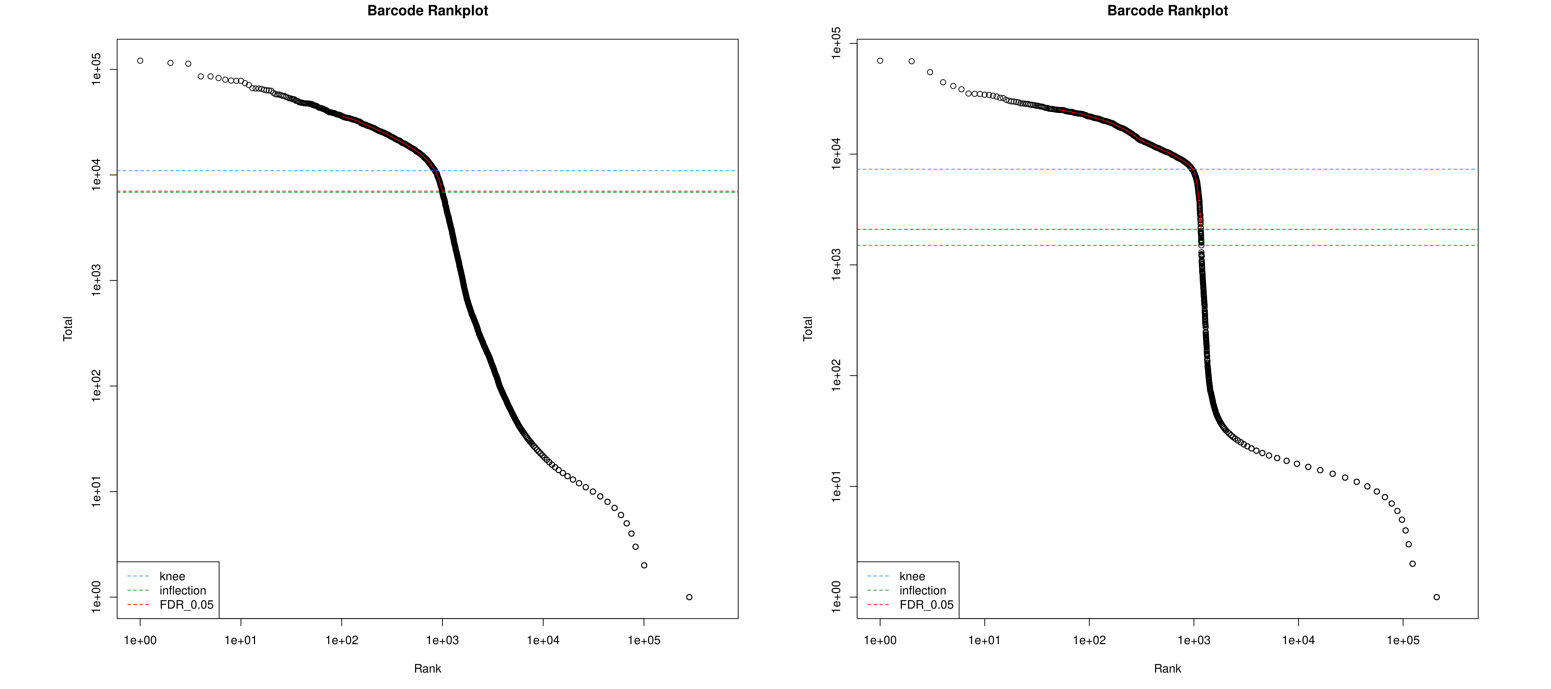
Brain_Tumor_3p(왼쪽)와 PBMC_1k_v3(오른쪽)의 바코드 랭크 플롯. 샘플 내 droplet들을 filtering 이전 UMI 수(y축)에 따라 정렬하여 시각화한 것으로, 붉은 점선(FDR 0.05) 아래의 droplet은 empty로 간주되어 제거됨 점선은 실제 세포와 비세포 바코드를 구분하기 위한 knee(카운트가 급격히 감소하는 지점), inflection(보다 보수적인 기준), FDR 0.05(emptyDrops에서 계산된 통계적 기준)을 나타냄
-

dropletUtils filterCells 결과 폴더 구조
scanpy_applyQCthresholds
Scanpy applyQCthreshold는 QC 지표가 계산된 AnnData object(.h5ad 파일)를 불러와 사용자가 지정한 QC 범위에 따라 세포를 필터링합니다. 이 과정에서는 총 UMI 수, 세포별 발현 유전자 수, 미토콘드리아 유전자 비율 등을 기준으로 낮은 품질의 세포를 제거할 수 있습니다. 필터링된 세포는 샘플별 개별 AnnData로 저장하거나 모든 샘플을 통합한 AnnData로 저장할 수 있습니다. 임계값 적용 이후 각 샘플의 QC 지표(n_genes_by_counts, total_counts, pct_counts_mt) 분포는 히스토그램과 산점도 플롯으로 시각화되며, 이를 통해 사용자는 지정한 QC 기준이 적절하게 low-quality 세포를 제거하고, 고품질 세포를 유지했는지 직관적으로 검토할 수 있어 이후 분석의 신뢰성을 높일 수 있습니다.
주요사항
- lower_log1p_total_counts, lower_log1p_n_genes_by_counts, upper_pct_counts_mito 등을 직접 지정하여 데이터 특성과 연구 목적에 맞게 품질 기준을 조정할 수 있습니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_applyQCthresholds.py input_dir = ./scanpy_plotqcmetrics/output \ output_dir = ./scanpy_applyqcthresholds/output \ lower_log1p_total_counts 0,0 \ lower_log1p_n_genes_by_counts 0,0 \ upper_pct_counts_mito 100,100 \ plotting_only False \ concat_samples True
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_plotqcmetrics/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_plotQCmetrics 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_applyqcthresholds/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | lower_log1p_total_counts | 0 | 샘플별 log1p_total_counts의 하한값으로, 이 값 이하의 세포를 low-quality로 필터링하는 기준값. 여러 샘플일 경우 쉼표로 구분 | |
| Option | String | lower_log1p_n_genes_by_counts | 0 | 샘플별 log1p_n_genes_by_counts의 하한값으로, 이 값 이하의 세포를 low-quality로 필터링하는 기준값. 여러 샘플일 경우 쉼표로 구분 | |
| Option | String | upper_pct_counts_mito | 100 | 샘플별 pct_counts_mito의 상한값으로, 이 값 이상인 세포를 low-quality로 필터링하는 기준값. 여러 샘플일 경우 쉼표로 구분 | |
| Option | Boolean | plotting_only | False | True: 시각화 파일만을 저장, False: 시각화 파일 및 QC를 마친 AnnData를 저장 | |
| Option | Boolean | concat_samples | True | True: 여러 샘플에 대한 AnnData를 병합하여 저장 False: 각각의 샘플에 대한 AnnData를 저장 |
결과
-
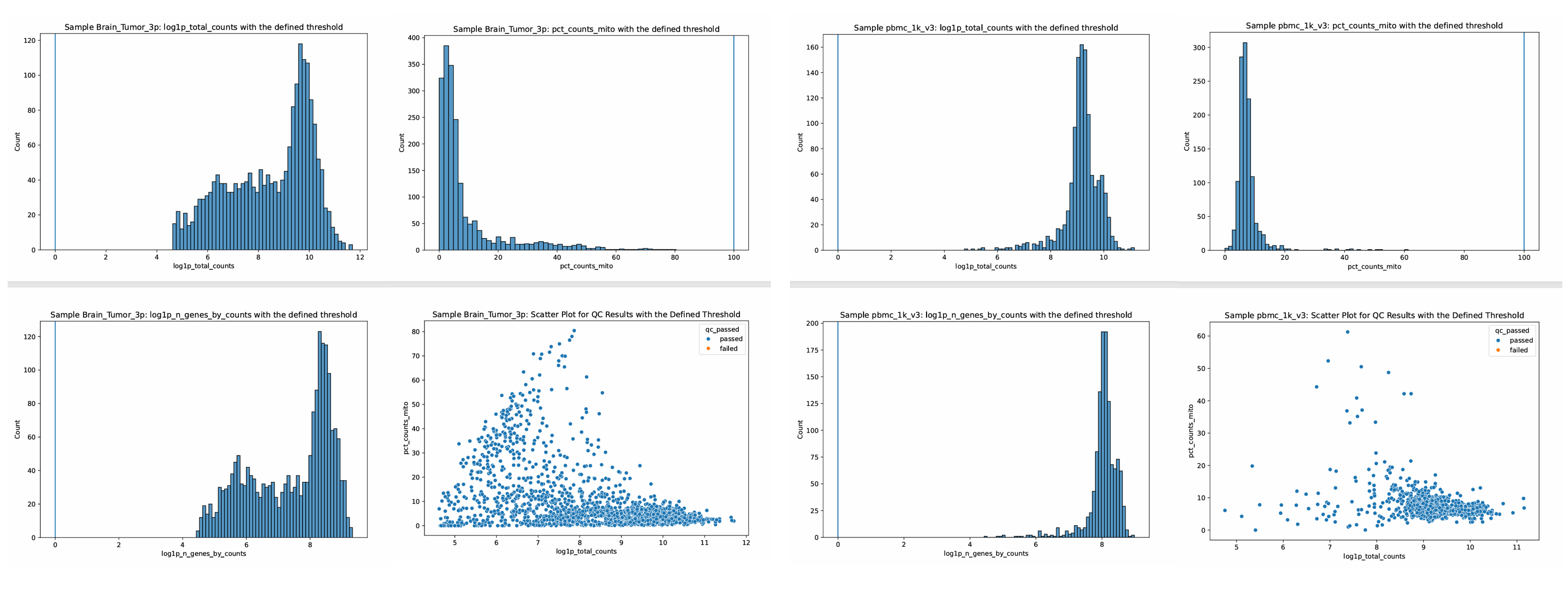
Brain_Tumor_3p(왼쪽)와 PBMC_1k_v3(오른쪽)의 히스토그램 및 산점도 플롯. 설정한 QC 임계값을 적용한 후 남은 세포들의 품질을 시각화하여, 사용자가 지정한 기준에 따라 low-quality 세포가 효과적으로 제거되었는지 확인 가능
-
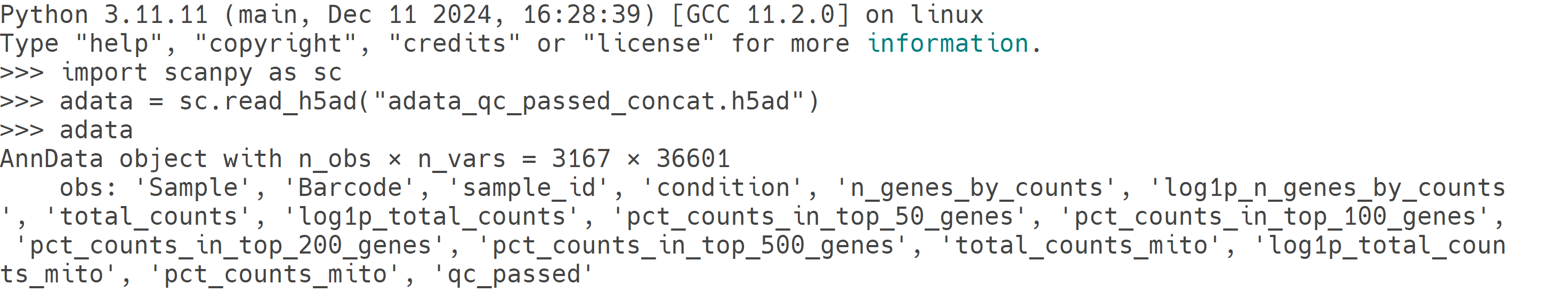
adata_qc_passed _concat.h5ad 파일 내용. 3,176개의 세포와 36,601개의 유전자에 대한 발현 데이터를 포함한 AnnData 객체로, 샘플 정보, 세포 품질 지표, 유전자 수준의 발현 통계값 및 QC 통과 여부를 포함함
-

scanpy applyQCthresholds 결과 폴더 구조
scanpy_normalize
세포의 유형이나 크기에 따라 세포당 총 RNA 수는 크게 달라질 수 있으며, 이러한 차이는 scRNA-seq 데이터 분석에서 발현량 편향이나 downstream 분석 오류를 유발할 수 있습니다. 이러한 문제를 보정하기 위해 Bio-Express에서는 Scanpy에서 제공하는 count depth scaling 방법을 사용하여 각 세포의 총 UMI 수를 기준으로 발현량을 조정합니다. 모든 발현량 조정이 끝난 후에는 각 값에 1을 더하고 자연로그(log1p)를 취하여 발현량 분포의 왜도(skewness)를 완화하고, 극단값에 의한 영향을 줄입니다. 변환된 데이터는 AnnData 객체의 기본 위치(X) 또는 사용자가 지정한 layer에 저장되며, 이후 normalization이 필요한 분석 단계에서 그대로 활용할 수 있습니다. 이를 통해 서로 다른 세포 간의 발현량 비교를 보다 정확하게 수행할 수 있습니다.
주요사항
- scanpy.pp.normalize_total() 함수에서 target_sum 옵션은 각 세포마다 총 발현량(UMI 총합)을 동일하게 정규화하는 기준값을 의미합니다. 이 옵션을 잘못 설정하면 downstream 분석에 영향을 줄 수 있습니다. target_sum 값은 분석 목적에 따라 1e4는 scRNA-seq에서 표준적으로 가장 많이 사용되며, 1e6값은 bulk RNA-seq과 같은 TPM 스타일의 정규화 방식과 유사합니다.
실행 명령어 예시
python scanpy_normalize.py input_dir = ./scanpy_applyqcthresholds/output \ output_dir = ./scanpy_normalize/output \ target_sum = 10000 \ exclude_highly_expressed = FALSE \ key_added = None \ layer = None
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_applyqcthresholds/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_applyQCthresholds 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_normalize/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Float | target_sum | 10000 | Normalization 후 각 세포 내 총 UMI count가 target_sum 값과 동일하게 함. None인 경우normalization 이전 각 세포 내 의 총 UMI count의 중앙값과 같도록 함 | |
| Option | Boolean | exclude_highly_expressed | FALSE | 각 세포의 normalization 계산 시, 한 세포에서 너무 높은 비율로 발현되는 유전자를 제외할지 여부. 제외되지 않은 유전자 합은 target_sum이 됨 | |
| Option | String | key_added | None | 계산된 normalization factor를 저장할 adata.obs 내 필드 이름 | |
| Option | String | layer | None | Normalization을 수행할 layer 지정. None이면 기본 X를 normalization함 |
결과
-

adata_normalized.h5ad 파일 내용. Normalization 후, 모든 발현량에 1을 더하고 자연로그(log1p)를 취한 발현값이 포함된 AnnData 객체
scanpy_findHVGs
단일 세포 발현 매트릭스는 수많은 세포와 유전자를 포함하기 때문에 매우 높은 차원을 가지며, 직접적인 분석은 계산량이 크고 해석이 어려울 수 있습니다. 이를 완화하기 위해, 전체 세포 집단에서 세포 간 발현 차이를 잘 설명할 수 있는 highly variable gene (HVG)를 선별하여 데이터 차원을 감소시키는 것이 일반적입니다. scanpy_findHVG는 log-normalized expression matrix를 기반으로, 각 유전자의 평균 발현량 대비 분산이 높은 유전자들을 HVG로 선정합니다. 선정된 HVG는 downstream 분석에서 주로 사용되며, 결과물로는 HVG가 포함된 AnnData 객체, HVG 리스트, 그리고 mean-dispersion plot이 제공됩니다.
주요사항
- 반드시 Normalization (sc.pp.normalize_total() 및 sc.pp.log1p() ) 수행 후에 실행해야 합니다.
- subset=True 옵션 설정 시 선택된 HVG만 남고 나머지 유전자는 AnnData에서 삭제됩니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_findHVGs.py input_dir = ./scanpy_normailize/output \ output_dir = ./scanpy_findhvgs/output \ flavor =seurat \ n_top_genes = none \ layer = none \ min_mean = 0.0125 \ max_mean = 3 \ min_disp = 0.5 \ max_disp = Inf \ span = 0.3 \ n_bins = 20 \ subset = FALSE
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_normailize/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_normalize 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_findhvgs/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | flavor | seurat | HVG를 식별하는 방법 선택. Seurat은 dispersion의 cutoff 사용, Cell Ranger는 dispersion 기반 상위 n개의 유전자를 선별함. [가능한 값: "seurat", "cell_ranger"] | |
| Option | Integer | n_top_genes | none | 유지할 상위 HVG의 수. | |
| Option | String | layer | none | HVG를 계산의 대상이 될 AnnData 내 발현량의 layer. None인 경우 AnnData의 X를 사용함. (추후 PCA가 X를 이용하여 진행되므로, [layer] 별도 설정을 권장하지 않음) | |
| Option | Float | min_mean | 0.0125 | HVG 선정에 사용되는 발현량의 mean 최소값 | |
| Option | Float | max_mean | 3 | HVG 선정에 사용되는 발현량의 mean 최대값 | |
| Option | Float | min_disp | 0.5 | HVG 선정에 사용되는 발현량의 dispersion의 최소 값 | |
| Option | Float | max_disp | Inf | HVG 선정에 사용되는 발현량의 dispersion의 최대 값 | |
| Option | Float | span | 0.3 | loess 모델 fitting에서 dispersion을 추정할 때 사용되는 데이터(세포)의 비율 | |
| Option | Integer | n_bins | 20 | 평균 발현량을 binning할 때 사용할 구간 수. 각 구간에 대해 normalization이 수행됨. 특정 구간에 1개의 유전자만 포함되면, 그 유전자의 normalized dispersion은 1로 강제 설정됨 | |
| Option | Boolean | subset | FALSE | 전체 유전자가 아닌 HVG에 대한 AnnData를 저장함. (True인 경우 이후 분석 및 시각화에 유의 필요) |
결과
-
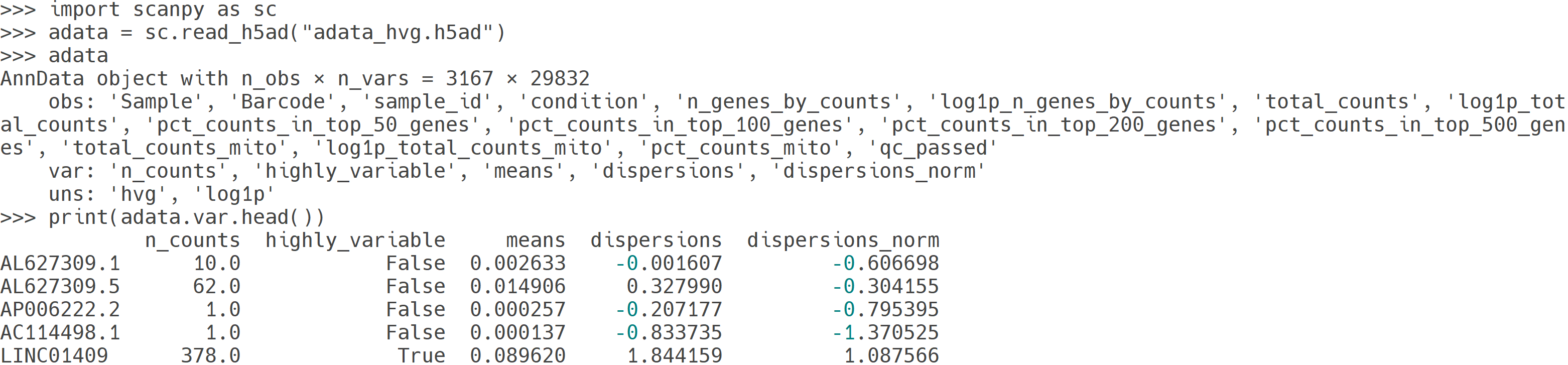
adata_hvg.h5ad 파일 내용. HVGs만 포함된 발현 데이터를 저장한 AnnData 객체. Normalized 결과와 비교하였을 때 'highly_variable', 'means', 'dispersions', 'dispersions_norm' 변수가 추가됨
-
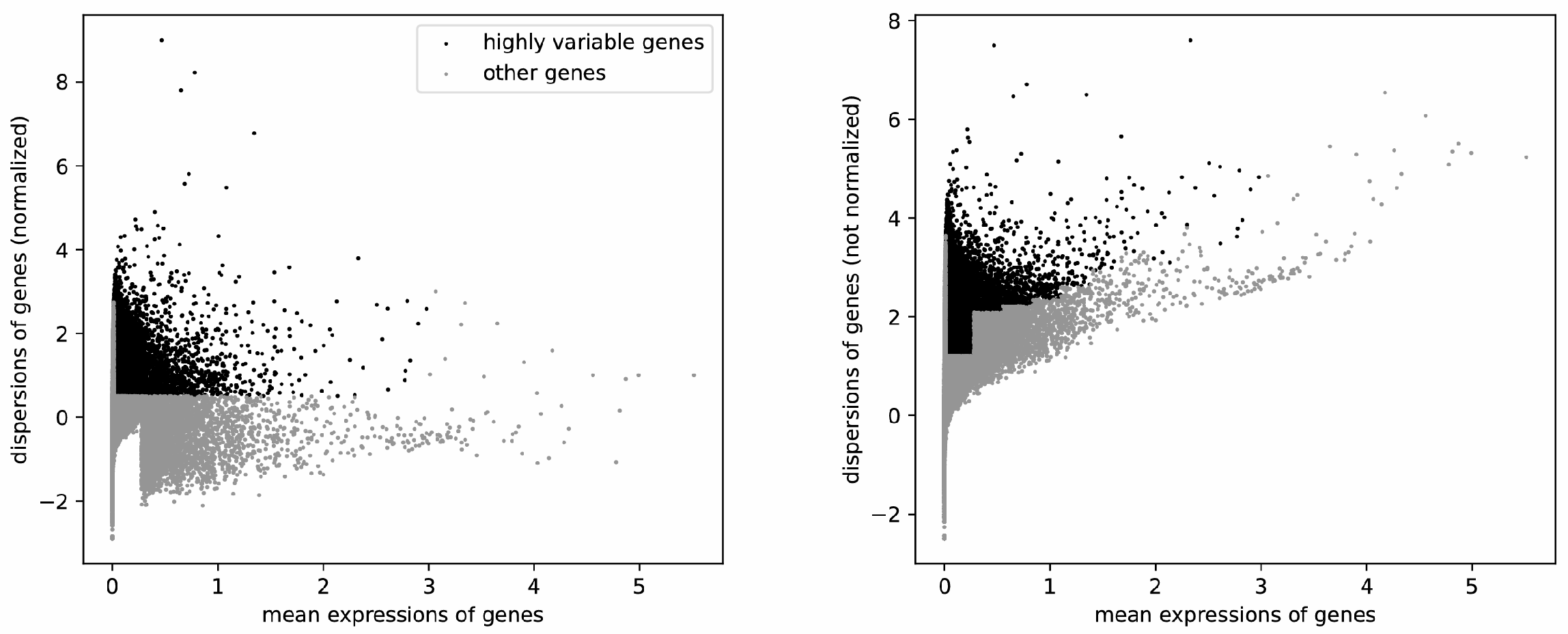
HVG scatter 플롯 내용. 전체 유전자에 대한 평균 발현량 대비 분산을 시각화한 그래프. HVG는 평균 대비 분산이 높아, plot 상에서 상단 또는 특정 cutoff 영역에 표시되므로 이를 통해 HVG 선별 과정이 적절했는지 직관적으로 확인 가능
-

hvg_list.csv 파일 내용. HVG로 선정된 유전자 목록을 담은 파일로 downstream 분석에서 차원 축소(PCA, UMAP 등)나 클러스터링에 사용됨
scanpy_scaleData
Log-normalization을 마친 AnnData 객체의 발현량을 바탕으로, 전체 세포 집단에서 각 유전자의 발현량을 Z-score 방식으로 표준화(z-scaling)합니다. 이 과정을 통해 유전자 간 발현량의 차이를 정규화하여, 특정 유전자가 PCA, UMAP, 클러스터링과 같은 downstream 분석에서 지나치게 큰 영향을 미치는 것을 방지합니다. 또한, 모든 유전자의 발현값이 동일한 척도로 변환됨으로써 분석 결과가 보다 공정하고 안정적으로 산출되며, 데이터의 변동성이 높은 유전자가 과도하게 결과에 영향을 주는 것을 막아 차원 축소 및 클러스터링 결과의 신뢰도를 높이는 데 기여합니다. 이 표준화된 데이터는 이후 분석에 최적화된 형태로 활용됩니다.
주요사항
- 극단치(outlier)는 Z-scaling에 영향을 줄 수 있어, 일반적으로 max_value=10으로 제한합니다. 너무 낮게 설정하면 고발현 유전자(HVGs)가 잘릴 수 있으므로 주의가 필요합니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_scaleData.py input_dir = ./scanpy_findhvgs/output \ output_dir = ./scanpy_scaledata/output \ zero_center = True \ max_value = None \ layer = None
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_findhvgs/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_findHVGs 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_scaledata/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Boolean | zero_center | True | False일 경우, zero-centering variable을 생략하여 sparse한 data(0이 많은 expression matrix)를 효율적으로 처리 가능 | |
| Option | Integer | max_value | None | Scaling 이후 [max_value] 이상의 값을 [max_value]로 치환함. None인 경우 치환하지 않음 | |
| Option | String | layer | None | Scaling의 대상이 될 AnnData 내 발현량의 layer. None인 경우 AnnData의 X를 사용함. (추후 PCA가 X를 이용하여 진행되므로, layer 별도 설정을 권장하지 않음) |
결과
-
.png)
adata_scaled.h5ad 파일 내용. 유전자의 발현값을 Z-score 방식으로 표준화(z-scaling)하여 저장한 AnnData 객체. HVG 선정 결과와 비교하였을 때 'mean', 'std' 변수가 추가됨
scanpy_runPCA
scanpy_runPCA는 고차원 유전자 발현 데이터를 저차원으로 축소해 주요 패턴을 추출하는 과정입니다. scRNA-seq 데이터는 차원이 높고 노이즈가 많기 때문에, PCA(Principal Component Analysis, 주성분 분석)를 통해 변동성이 큰 방향을 찾아 요약합니다. 주로 HVG(Highly Variable Genes)를 기반으로 수행하여 생물학적으로 의미 있는 변동성을 반영합니다. 이렇게 얻은 주성분은 데이터의 구조를 유지하면서 계산 효율성을 높이고, 이후 클러스터링이나 시각화(UMAP, t-SNE) 같은 분석의 기초로 활용됩니다.
주요사항
- PCA가 의미 있게 작동하려면 정규화, 로그 변환, HVG 선정, 스케일링 작업이 선행되어야 힙니다. 특히 스케일링을 하지 않으면 분산이 큰 유전자만 PCA에 과도하게 기여합니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_runPCA.py input_dir = ./scanpy_scaledata/output \ output_dir = ./scanpy_runpca/output \ svd_solver = auto \ n_comps = 50 \ zero_center = True \ chunked = False \ chunk_size = None \ random_state = 0
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_scaledata/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_scaleData 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_runpca/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Integer | n_comps | 50 | 계산할 principal component의 수. 1을 입력하면 계산 가능한 최소 차원으로 계산됨. | |
| Option | Boolean | zero_center | True | True: 공분산 행렬을 통해 표준 PCA를 계산; False: zero-centering을 하지 않고 TruncatedSVD 방식(scikit-learn 또는 dask-ml에서 제공)을 사용. 희소(sparse) 데이터일 때 메모리와 속도 면에서 훨씬 효율적임; None: 데이터가 희소인지 아닌지를 자동으로 판단하여 처리 | |
| Option | String | svd_solver | auto | 사용할 SVD solver 선택. 가능한 값: "arpack", "randomized", "auto", 기본값: "auto" ; Auto: scanpy가 자동으로 지정; Arpack: SciPy의 ARPACK wrapper (svds()) 사용하며 dask array에 대해 사용 불가; Randomized: Halko의 randomized 알고리즘 사용 | |
| Option | Boolean | chunked | False | True: sklearn IncrementalPCA나 dask-ml IncrementalPCA를 이용해 chunk_size 크기의 segment 단위로 PCA 수행 (자동으로 데이터의 평균을 0으로 만들고, random_seed와 svd_solver 설정을 무시); False: sklearn PCA나 dask-ml PCA를 이용해 full PCA 수행 | |
| Option | Integer | chunk_size | None | 각 chunk에 포함할 관측값의 수입니다. ([chunked]=True인 경우 필수) | |
| Option | Integer | random_state | 0 | 랜덤 시드 값 |
결과
-
.png)
Scree 플롯 내용. PCA 후 각 주성분이 설명하는 분산 비율을 보여주는 그래프로 x축은 주성분 번호, y축은 분산 비율이며, 주성분 개수를 결정할 때 참고
-
.png)
adata_scaled.h5ad 파일 내용. Z-score로 표준화된 발현 데이터를 기반으로 PCA(주성분 분석)를 수행한 결과를 저장한 AnnData 객체로, obsm['X_pca']에는 각 세포의 주성분 점수(PC scores)가, varm['PCs']에는 각 유전자의 주성분 로딩(PC loadings) 정보가 저장되어 있습니다. 또한 uns['pca']에는 분산 비율(explained variance ratio) 등 PCA 관련 요약 정보가 포함됨
-
.png)
PCA_embedding.csv 파일 내용. 각 세포(barcode)의 PCA 점수를 저장한 파일로, 이후 클러스터링이나 UMAP/t-SNE 시각화에 사용
scanpy_findNeighbors
findNeighbors는 scRNA-seq 발현량 데이터를 기반으로 세포 간 유사성을 평가하여 neighbor graph를 구축하는 과정입니다. 각 세포는 유전자 발현 패턴이 비슷한 이웃 세포들과 연결되며, edge에는 발현 패턴의 유사도에 따라 가중치가 부여됩니다. 이를 통해 각 세포에 대해 유사도가 높은 세포들을 효율적으로 선별할 수 있으며, 생성된 neighbor graph는 후속 군집 분석(clustering), 차원 축소(UMAP, t-SNE) 및 다양한 downstream 분석에서 세포 간 구조를 반영하는 기초 자료로 활용됩니다.
주요사항
- sc.pp.neighbors()는 주로 PCA 결과(obsm['X_pca'])를 사용하므로, PCA를 먼저 수행해야 합니다. PCA 없이 호출하면 adata.X로 계산되어 효율이 떨어지고 결과가 왜곡될 수 있습니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_findNeighbors.py input_dir = ./scnapy_runpca/output \ output_dir = ./scanpy_findNeighbors/output \ n_neighbors = 15 \ n_pcs = 50 \ use_rep = None \ knn = True \ method = umap \ metric = euclidean \ random_state = 0
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scnapy_runpca/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_runPCA 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_findNeighbors/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Integer | n_neighbors | 15 | 각 세포 주변을 살펴볼 이웃의 수로, 값이 클수록 전체 구조를, 작을수록 국소 구조를 반영함. knn=True이면 지정한 수만큼의 이웃만 고려하고, knn=False이면 Gaussian kernel을 이용해 이웃 거리에 따라 가중치를 부여 | |
| Option | Integer | n_pcs | 50 | Neighbor graph 계산에 사용할 주성분의 수. [n_pcs]=0일 경우 [use_rep]가 None이면 원본 데이터(.X)를 사용 | |
| Option | String | use_rep | None | Neighbor graph 계산에 사용할 데이터 지정. X: 원본 발현 데이터 사용; X_pca: PCA로 축소한 발현 데이터 사용; None: scanpy가 자동으로 결정 | |
| Option | Boolean | knn | True | True: 이웃의 수를 [n_neighbors]로 제한하기 위해 hard threshold를 사용함 (k-NN graph 고려); False: Gaussian kernel을 사용하여 [n_neighbors] 이웃보다 더 먼 이웃에 대해 낮은 가중치 할당 | |
| Option | String | method | umap | Connectivity를 계산할 때 사용할 방법 지정. 기본값: "umap"; 가능한 값: "umap", "gauss" | |
| Option | String | metric | euclidean | 데이터 간의 Distance를 계산하는 metric 명이나 함수 지정. 기본값: 'euclidean'; 가능한 값: 'cityblock', 'cosine', 'euclidean', 'l1', 'l2', 'manhattan', 'braycurtis', 'canberra', 'chebyshev', 'correlation', 'dice', 'hamming', 'jaccard', 'kulsinski', 'mahalanobis', 'minkowski', 'rogerstanimoto', 'russellrao', 'seuclidean', 'sokalmichener', 'sokalsneath', 'sqeuclidean', 'yule' | |
| Option | Integer | random_state | 0 | 랜덤 시드 값 |
결과
-
.png)
adata_scaled.h5ad 파일 내용. 세포 간 이웃 정보를 계산하여 저장한 AnnData 객체로, 각 세포의 neighbor graph 정보(obsp['connectivities'], obsp['distances'])가 포함됨
scanpy_runTSNE
scanpy_runTSNE는 scRNA-seq 데이터에 대해, UMAP 등장 이전에 사용하던 비선형적 차원 축소 방식인 t-stochastic neighbor embedding(t-SNE)를 수행합니다. t-SNE는 고차원 공간에서 각 점과 그 이웃 간의 거리를 보존하는 저차원 공간을 찾습니다. 이를 통해 데이터의 국지적인 구조를 유지함으로써, 복잡한 집단에서 많은 서로 다른 클러스터를 2차원상에서 분리되도록 embedding합니다. 때에 따라 scanpy_runUMAP 단계로 대체할 수 있습니다. (UMAP은 전반적인 데이터의 구조를 유지하지만, t-SNE는 국지 구조만을 유지한다는 단점을 가집니다.)
주요사항
- t-SNE는 확률 분포 기반 유사도를 보존하여 임베딩을 수행하며, 특히 클러스터 내부의 거리와 형태 해석에 신뢰성이 높습니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_runTSNE.py input_dir = ./scanpy_findNeighbors/output \ output_dir = ./scanpy_runtsne/output \ n_pcs = 50 \ use_rep = none \ perplexity = 30 \ early_exaggeration = 12 \ learning_rate = 1000 \ random_state = 0
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_findNeighbors/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_findNeighbors 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_runtsne/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Integer | n_pcs | 50 | t-SNE를 계산하기 위해 사용할 주성분의 수. [n_pcs]=0일 경우 [use_rep]가 None이면 원본 데이터(.X)를 사용 | |
| Option | String | use_rep | none | t-SNE 계산에 사용할 데이터 선택. X: 원본 발현 데이터 사용; X_pca: PCA로 축소한 발현 데이터 사용; None: scanpy가 자동으로 결정 | |
| Option | Integer | perplexity | 30 | 데이터 포인트가 고려하는 이웃 수 조절, 5~50 권장 | |
| Option | Integer | early_exaggeration | 12 | 초기 클러스터 간 거리 확대, 값이 클수록 간격 증가 | |
| Option | Integer | learning_rate | 1000 | 최적화를 위한 학습률. 보통 100과 1000 사이의 값을 권장. | |
| Option | Integer | random_state | 0 | 랜덤 시드 값 |
결과
-
adata_scaled.h5ad 파일 내용. t-SNE를 수행한 결과를 저장한 AnnData 객체로, 각 세포의 저차원 임베딩 정보(obsm['X_tsne'])가 포함됨
-

tSNE_embedding.csv 파일 내용. 각 세포(barcode)의 t-SNE 좌표를 저장한 파일로, 이후 클러스터링 또는 시각화에 사용됨
scanpy_runUMAP
runUMAP은 앞서 계산한 scRNA-seq 데이터의 neighborhood graph를 기반으로, 비선형 차원 축소 기법인 UMAP(Uniform Manifold Approximation and Projection)을 수행합니다. 이 과정은 고차원 유전자 발현 데이터를 저차원 공간으로 변환하면서, 데이터의 전반적인 구조와 세포 간 관계를 최대한 유지하도록 설계되었습니다. UMAP을 통해 얻은 임베딩은 각 세포의 위치 정보를 2차원 또는 3차원 공간에 나타낼 수 있으며, 이를 바탕으로 클러스터링, 시각화, 군집 간 관계 분석 등에 활용할 수 있습니다. 경우에 따라, UMAP 대신 scanpy_runTSNE 단계를 사용하여 국지적 구조를 강조한 비선형 임베딩을 수행할 수도 있습니다.
주요사항
- UMAP은 sc.pp.neighbors()에서 계산한 KNN 그래프(connectivities)를 입력으로 사용하여, 고차원 데이터의 지역 구조(local structure)와 전역 구조(global structure)를 모두 보존하도록 저차원 공간에 임베딩 합니다. 이때 점(세포) 간 거리는 유전자 발현 패턴의 유사성을 반영하며, 가까운 점은 유사한 세포, 먼 점은 발현 패턴이 다른 세포를 의미합니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_runUMAP.py input_dir = ./scanpy_runtsne/output \ output_dir = ./scanpy_runumap/output \ min_dist = 0.5 \ spread = 10 \ n_components = 2 \ maxiter = none \ alpha = 1.0 \ gamma = 1.0 \ negative_sample_rate = 5 \ init_pos = random \ random_state = 0 \ a = none \ b = none
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_runtsne/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_findNeighbors 또는 scanpy_runTSNE 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_runumap/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Float | min_dist | 0.5 | 임베딩된 점(세포)들 간의 최소 거리. 작은 값은 더 밀집된 cluster를 만들고, 클수록 점들이 더 고르게 분포됨. [spread]와의 조합으로 점의 밀집도를 조절함. umap-learn 패키지에서 기본값은 0.1. | |
| Option | Float | spread | 1.0 | [min_dist]와 함께 클러스터링의 밀집 정도를 지정, 값이 클수록 넓게 퍼짐 | |
| Option | Integer | n_components | 2 | 임베딩할 차원 수 | |
| Option | Integer | maxiter | none | 최적화를 위한 반복 횟수(epochs). 기존 UMAP에서는 [n_epochs]로 표기됨. | |
| Option | Float | alpha | 1.0 | 임베딩 최적화의 초기 학습률 | |
| Option | Float | gamma | 1.0 | 임베딩 최적화 시 negative sample에 적용되는 가중치. 값이 1보다 크면 negative sample에 더 큰 가중치가 부여됨 | |
| Option | String | negative_sample_rate | 5 | 저차원 임베딩을 최적화할 때, 유사하다고 판단된 세포 쌍(positive edge) 하나당 사용할 유사하지 않은 세포 쌍(negative edge)의 수 | |
| Option | String | init_pos | random | 임베딩의 초기화 방법. 기본값: 'random'; 가능한 값: 'paga', 'spectral', 'random' | |
| Option | Integer | random_state | 0 | 랜덤 시드 값 | |
| Option | Float | a | none | 임베딩을 제어하는 더 구체적인 parameter. None일 경우 [min_dist]와 [spread]에 의해 자동으로 설정 | |
| Option | Float | b | none | 임베딩을 제어하는 더 구체적인 parameter. None일 경우 [min_dist]와 [spread]에 의해 자동으로 설정 |
결과
-
adata_scaled.h5ad 파일 내용. UMAP을 수행한 결과를 저장한 AnnData 객체로, 각 세포의 저차원 임베딩 정보(obsm['X_umap'])가 포함됨
-

umap_embedding.csv 파일 내용. 각 세포(barcode)의 UMAP 좌표를 저장한 파일로, 이후 클러스터링 또는 시각화에 사용됨
scanpy_findClusters
scanpy_findClusters 단계는 앞서 계산된 scRNA-seq 데이터의 neighbor graph(FindNeighbors 단계 결과)를 기반으로, 각 세포를 유사한 발현 패턴을 가진 그룹으로 묶는 클러스터링 과정을 수행합니다. 이때 Leiden 또는 Louvain 알고리즘을 사용하여 세포 집단을 자동으로 식별하며, 알고리즘은 세포 간 연결 강도와 구조를 고려하여 최적의 군집을 형성합니다. 클러스터 번호는 세포 수가 많은 그룹부터 0, 1, 2… 순으로 할당되며, 결과는 AnnData 객체의 obs에 저장되어 추후 시각화, 차원 축소(UMAP/t-SNE), 또는 집단 간 차이 분석에 활용할 수 있습니다. 이 과정은 scRNA-seq 데이터에서 세포 유형 및 상태를 탐색하는 핵심 단계입니다.
주요사항
- sc.tl.leiden()과 sc.tl.louvain()은 이웃 그래프를 기반으로 세포 간 유사도에 따라 군집을 나누는 방법입니다. Louvain은 빠르지만 분할이 불안정할 수 있고, Leiden은 이를 개선하여 더 안정적이고 해석력이 높아 Scanpy에서 주로 권장됩니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_findClusters.py input_dir = ./scanpy_runumap/output \ output_dir = ./scanpy_finclusters/output \ algo = leiden \ resolution = 1.0 \ use_weights = True \ random_state = 0 \ directed = none \ flavor = vtraag \ leiden_n_iter = -1
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_runumap/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_runTSNE 또는 scanpy_runUMAP 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_finclusters/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | algo | leiden | Clustering에 사용할 알고리즘으로 선택한 알고리즘에 따라 일부 파라미터의 기본값이 달라짐. 기본값: "leiden"; 가능한 값: "louvain", "leiden" | |
| Option | Float | resolution | 1.0 | Clustering의 세밀도를 조절하는 파라미터. 높을수록 더 많은 cluster를 생성 | |
| Option | Boolean | use_weights | True | k-NN graph의 가중치의 사용 여부. True일 경우 edge weight를 계산에 사용 | |
| Option | Integer | random_state | 0 | 최적화를 위한 초기값 | |
| Option | Boolean | directed | none | adjacency matrix을 directional graph로 해석할지 여부 설정 | |
| Option | String | flavor | vtraag | (Louvain) 클러스터링 계산을 위한 패키지 선택. 기본값: "vtraag":, 가능한 값: "vtraag", "igraph" | |
| Option | Integer | leiden_n_iter | -1 | (Leiden) 알고리즘을 몇 번 반복할지 설정. - 2 이상의 양수: 총 반복 횟수 - -1: 알고리즘이 최적의 클러스터링에 도달할 때까지 실행 |
결과
-
adata_scaled.h5ad 파일 내용. 클러스터링을 수행한 결과를 저장한 AnnData 객체로, 세포별 클러스터 정보(obs['leiden'])가 포함됨
-

cluster_info_leiden.csv 파일 내용. 각 세포에 대해 할당된 Leiden 클러스터 정보를 담은 표
scanpy_scatterPlot
앞서 계산된 UMAP, t-SNE, 혹은 PCA 임베딩 결과를 활용하여 모든 세포에 대한 산점도를 시각화합니다. 이때 각 점은 하나의 세포를 나타내며, 샘플 정보나 findClusters 단계에서 얻은 클러스터 번호에 따라 색상 또는 모양으로 구분할 수 있습니다. 이러한 산점도는 군집 구조 확인을 넘어, 개별 세포 수준에서 특정 유전자의 발현 패턴을 탐색하는 데도 유용합니다. 예를 들어, raw count, log-normalized, scaled 등 다양한 데이터 변환 방식에 따른 발현 강도를 색상으로 표시하면 특정 유전자가 집단 특이적으로 발현되는지, 혹은 전반적으로 균일하게 분포하는지를 효과적으로 파악할 수 있습니다.
주요사항
- 시각화 하고자 하는 적절한 차원 축소방식을 선택해야 합니다. PCA는 전반적 구조 파악에, t-SNE는 국지적 구조 보존에, UMAP은 국지적·전역적 구조를 모두 반영하며 재현성과 속도가 뛰어나 최근 가장 널리 활용됩니다. 시각화 시 유전자 발현값은 raw, log-normalized, scaled 등 데이터 변환 수준에 따라 다르게 표현될 수 있으므로 적절한 표현 방식을 고려해야 합니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_scatterPlot.py input_dir = ./scanpy_findcluseter/output \ output_dir = ./scanpy_scatterPlot/output \ basis = umap \ layer = scaled \ color_keys = sample_id \ annotated = False \ edges = False \ edges_width = 0.1 \ edgens_color = grey \ legend_loc = "right maring" \ legend_fontsize = none \ size = none \ save = .pdf
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_findcluseter/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_findCluster, scanpy_BBKNNbatchCorrection, scanpy_annotateClusters, scanpy_runCellTypist 등의 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_scatterPlot/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | Boolean | annotated | False | 세포 유형의 라벨링이 완료된 AnnData 객체(adata_cellType_annotated.h5ad)를 불러올지 여부. 기본값으로는 'adata_scaled.h5ad'을 로드하며, 이 경우 세포 유형은 시각화할 수 없음 | |
| Option | String | basis | umap | 시각화에 사용할 임베딩 선택. 기본값: "umap"; 가능한 값: "umap", "tsne", "pca" | |
| Option | String | color_keys | sample_id | 시각화할 색상을 기준으로 사용할 이름 (runCellRanger_count의 [add_condition]을 통해 설정한 condition 이름도 가능함). 예시로 'leiden' 또는 'leiden,condition', runCellTypist를 이용해 세포 유형을 부여한 경우 'CellTypist'를 포함 가능 | |
| Option | String | layer | scaled | 시각화할 AnnData의 발현 layer 선택. 가능한 값: "raw", "normalized", "scaled" | |
| Option | Boolean | edges | False | 세포 간 edge를 표시 여부 | |
| Option | Float | edges_width | 0.1 | edge의 선 두께 | |
| Option | String | edges_color | grey | edge의 색상, 기본값: "grey" | |
| Option | String | legend_loc | "right maring" | 범례 위치. 가능한 값: "on data", "right margin", None 또는 matplotlib legend 위치 키워드 가능 | |
| Option | Float | legend_fontsize | none | 범례 글꼴의 크기 (pt 단위) | |
| Option | Float | size | none | 점의 크기. None일 경우 자동으로 120000 / n_cells로 계산. | |
| Option | String | save | 출력할 그림 파일의 확장자 지정. 기본값: ".pdf" |
결과
-
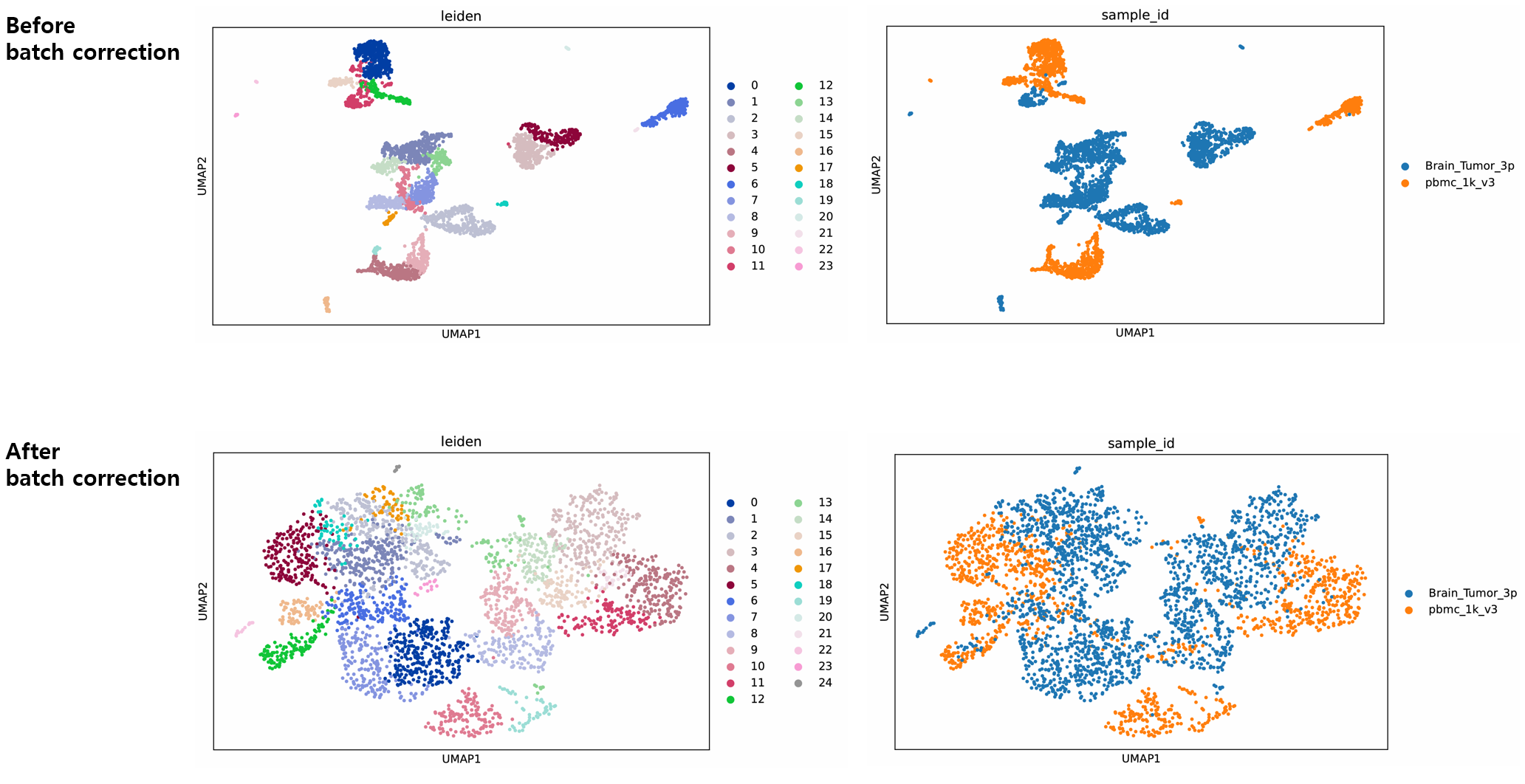
산점도 플롯. scRNA-seq 데이터의 UMAP 결과로, 각 점은 하나의 세포를 나타내고 leiden 클러스터링, sample_id에 따라 서로 다른 색상으로 구분됨. 위는 Batch correction 전, 아래는 Batch correction 이후 결과임
scanpy_BBKNNbatchCorrection
scRNA-seq 데이터는 기술적 요인(technical factors)의 영향을 받아 샘플 간 실제 생물학적 변이를 가릴 수 있으며, 이로 인해 연구 결과에 부정적인 영향을 미칠 수 있습니다. 대표적인 기술적 요인 중 하나인 batch effect는 샘플이 서로 다른 그룹이나 배치에서 처리될 때 발생할 수 있습니다. 이러한 영향을 최소화하기 위해 샘플을 동일한 날짜에 처리하고, 동일한 프로토콜과 장비를 사용하는 방법이 권장됩니다. 또한 batch effect correction을 위한 다양한 기법과 패키지(Harmony, MNN, ComBat 등)가 개발되어 있으며, Bio-Express에서는 Scanpy에 탑재된 BBKNN을 활용하여 batch effect correction 기능을 제공합니다. Batch correction 이후 scanpy_findNeighbors부터 scanpy_scatterPlot까지의 과정을 다시 수행해야 하며, 이를 통해 보정된 데이터 기반의 군집화 및 시각화 결과를 확인할 수 있습니다.
주요사항
- use_rep='X_pca'가 기본값이므로, PCA (sc.tl.pca())를 선행하여 AnnData 객체 내 obsm['X_pca']가 반드시 존재해야 합니다.
- 배치 정보 조건으로 batch_key가 adata.obs 내에 존재해야 하며, 최소 2개 이상의 배치 값이 필요합니다.
실행 명령어 예시
python scanpy_BBKNNbatchCorrection.py --input_dir /path/to/input_dir --output_dir /path/to/output_dir --batch_key sample_id --use_rep X_pca --n_pcs 50
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | /path/to/input_dir | 입력 파일이 저장된 디렉토리 경로로, PCA 결과를 포함하는 scanpy_runPCA, scanpy_Neighbors, scanpy_runTSNE, scanpy_runUMAP, scanpy_findClusters 등의 결과 폴더임 | |
| Output | Folder | output_dir | /path/to/output_dir | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | batch_key | sample_id | adata.obs 내에서 서로 다른 배치를 구분하는 컬럼 이름 | |
| Option | String | use_rep | X_pca | 이웃을 찾기 위해 사용할 데이터 선택. 기본값: "X_pca" | |
| Option | Integer | n_pcs | 50 | 분석에 사용할 차원 수 (PCA의 경우 주성분 수) | |
| Option | Boolean | approx | True | True: 가까운 세포를 찾기 위하여 annoy 또는 PyNNDescent 사용 (큰 데이터셋에서 실행 시간 단축, Batch correction 증가) | |
| Option | Boolean | use_annoy | True | [approx]=True일 때만 사용됨. True: annoy를 사용하여 neighbor finding 수행; False: pyNNDescent 사용 | |
| Option | String | metric | euclidean | 사용할 distance metric. Neighbor algorithm에 따라 옵션이 달라짐. 기본값:"euclidean"; Annoy의 가능한 값: 'angular', 'euclidean'; PyNNDescent의 가능한 값: 'euclidean', 'manhattan', 'hamming', 'l2', 'sqeuclidean', 'taxicab', 'l1', 'chebyshev', 'linfinity', 'linfty', 'linf', 'minkowski', 'seuclidean', 'standardised_euclidean', 'wminkowski' | |
| Option | Integer | neighbors_within_batch | 3 | 각 배치에 대해 몇 개의 상위 이웃을 report할지 설정 | |
| Option | Integer | trim | none | 각 세포의 이웃을 상위 [trim]개로 제한하여 클러스터링을 명확하게 함. 값이 작을수록 배치효과가 남고 population이 독립적으로 간주됨. None이면 [neighbors_within_batch] 값에 데이터 내 batch 수를 곱한 값의 10배로 자동 설정 | |
| Option | Integer | anny_n_trees | 10 | (Annoy) Annoy forest에서 생성할 tree의 수. 클수록 정확도가 올라가지만 실행 시간 증가 | |
| Option | Integer | pynndescent_n_neighbors | 30 | (pyNNDescent) 근사 그래프를 만들 때 고려할 이웃의 수. 클수록 정확도가 올라가지만 실행 시간 증가 | |
| Option | Integer | pynndescent_random_state | 0 | (pyNNDescent) 그래프 생성 시 사용할 랜덤 시드 값 | |
| Option | Boolean | use_faiss | True | [approx]=False, [metric]=“euclidean”일 때, neighbor 계산에 faiss를 사용하여 약간의 성능 향상 | |
| Option | Float | set_op_mix_ratio | 1.0 | UMAP connectivity 계산을 위한 파라미터. 0과 1 사이의 실수 | |
| Option | Integer | local_connectivity | 1 | UMAP connectivity 계산을 위한 파라미터, 각 세포에 대해 몇 개의 이웃이 완전히 연결된 것으로 가정되는지 설정 |
결과
-
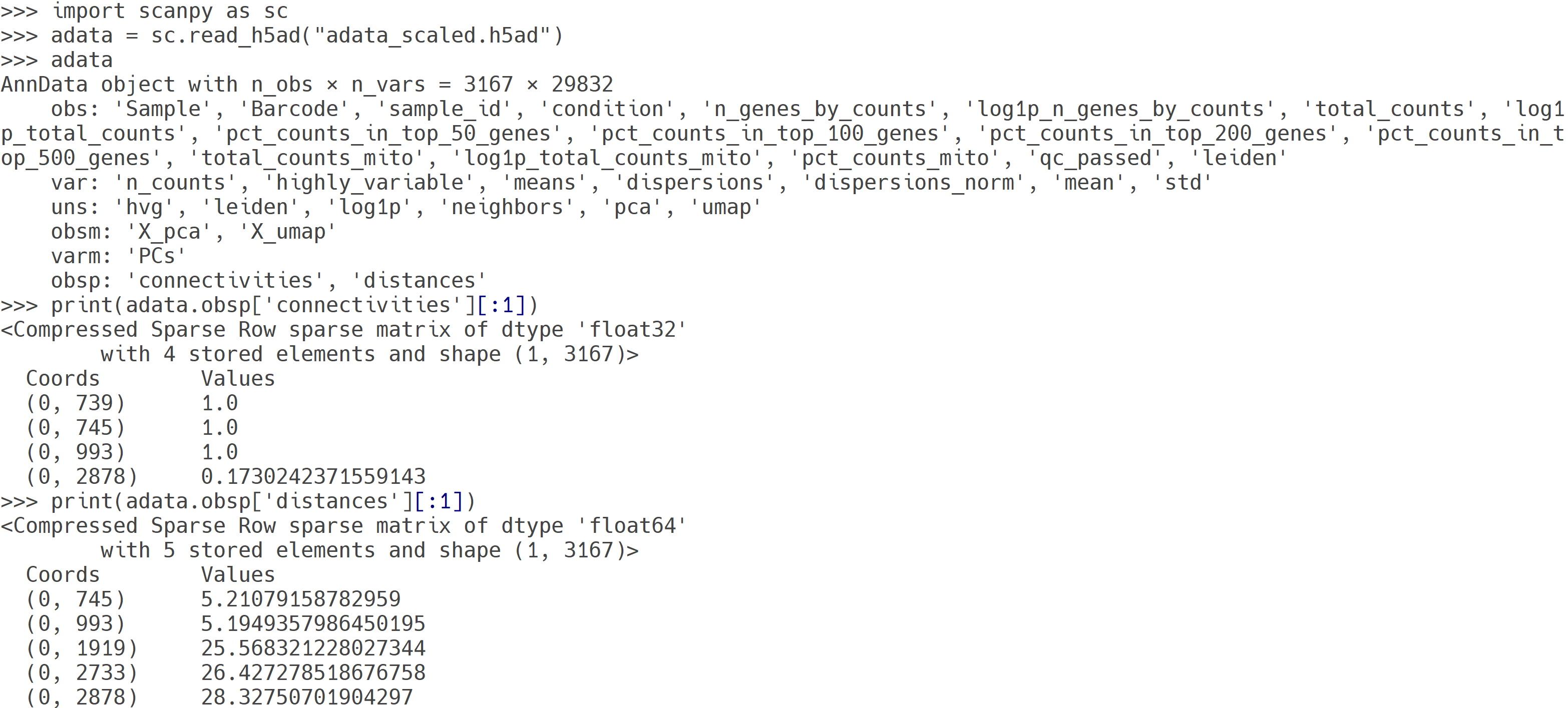
adata_scaled.h5ad 파일 내용. 세포 간 이웃 정보를 배치별로 균등하게 계산하여 저장한 AnnData 객체로, 원본 발현 데이터(.X)는 변하지 않지만, 각 세포의 neighbor graph 정보(obsp['connectivities'], obsp['distances'])는 배치 보정된 결과가 반영됨
scanpy_annotateClusters
Scanpy에서는 연구자가 특정 마커 유전자의 발현 패턴을 기준으로 직접 클러스터에 세포 유형을 부여할 수 있습니다. 예를 들어 T세포 마커(CD3D, CD3E), B세포 마커(MS4A1), 대식세포 마커(LYZ) 등의 발현을 확인하여 해당 클러스터가 어떤 세포 집단인지 판단합니다. 이후 클러스터 번호(예: 0, 1, 2…)에 연구자가 정의한 세포 유형 라벨을 매핑해 adata.obs['celltype'] 같은 새로운 열에 저장할 수 있습니다. 이 방법은 자동 주석 도구 대비 해석자의 전문 지식을 반영할 수 있어, 데이터 특성에 맞는 맞춤형 주석 작업이 가능하다는 장점이 있습니다.
주요사항
- 단일세포 데이터에서 세포들을 정규화(sc.pp.normalize_total)하고 로그 변환 후, 고변이 유전자를 선택해 PCA·neighbor 계산·UMAP 시각화와 함께 Leiden(또는 Louvain) 알고리즘으로 군집을 형성한 뒤(adata.obs['leiden']), 각 클러스터의 마커 유전자를 확인(sc.tl.rank_genes_groups)하거나 참조 데이터(sc.tl.ingest, CellTypist 등)를 이용해 세포 유형을 주석(adata.obs['cell_type'])하는 과정이다. 분석 시 이중세포 및 미토콘드리아 비율을 고려한 QC 필터링, 배치 효과 보정(sc.pp.combat, bbknn)을 수행하고, 해석에 사용된 파라미터와 라벨 정보를 함께 .h5ad로 저장하는 것이 중요하다.
입력 데이터 예제
실행 명령어 예시
python scanpy_annotateClusters.py input_dir = ./scanpy_findcluseter/output \ hvg_path = ./scanpy_findhvgs/output \ output_dir = ./scanpy_annotateclusters/output \ query_obs = leiden \ query_cat = 0,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 \ append_obs = celltype \ append_cat = T,T,T,Macrophages,Monocytes,T,Epithelial,Epithelial,Macrophages,Macrophages,B,Monocytes,NK,Macrophages,Macrophages,Macrophages,MAIT,Fibroblasts,Fibroblasts,B,Fibroblasts,Monocytes,T,Fibroblasts,Fibroblasts \ append_cat_order = none \
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | String | input_dir | ./scanpy_findcluseter/output | 입력 파일이 저장된 디렉토리 경로로, PCA 결과를 포함하는 scanpy_findClusters, scanpy_BBKNNbatchCorrection 등의 결과 폴더임 | |
| Input | String | hvg_path | ./scanpy_findhvgs/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_findHVGs 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_annotateclusters/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | query_obs | leiden | 기준이 되는 adata.obs의 컬럼 이름을 지정 | |
| Option | String | query_cat | none | 선택할 카테고리 값을 쉼표로 지정 (예시: 1,2,3,4) | |
| Option | String | append_obs | celltype | 새로운 주석 정보를 추가할 adata.obs의 컬럼 이름을 지정 | |
| Option | String | append_cat | none | 새롭게 추가할 카테고리 값을 쉼표로 지정 (예시: T-cell,B-cell,Myeloid,Myeloid) | |
| Option | String | append_cat_order | none | pandas.Categorical 형식으로 저장할 때 사용할 카테고리 순서를 쉼표로 구분해 지정합니다. 중복되지 않은 값만 입력해야 함 |
결과
-
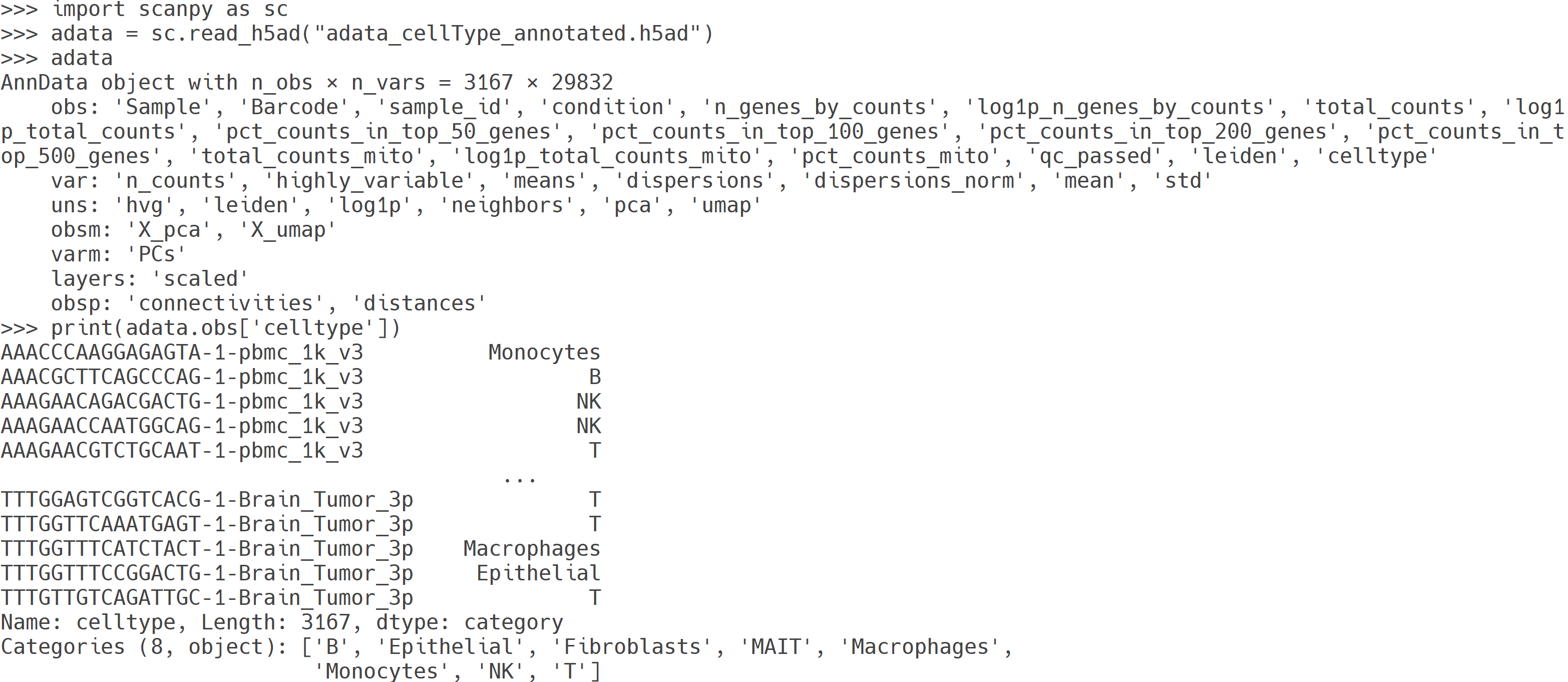
adata_cellType_annotated 파일 내용. 각 클러스터(leiden 등)를 기준으로, 연구자가 직접 세포 유형을 지정하여 주석(annotation)을 추가한 AnnData 객체로, 각 세포의 발현 데이터와 클러스터 정보뿐만 아니라, 사용자가 클러스터별로 지정한 세포 유형 정보(obs['celltype'])를 포함
scanpy_runCellTypist
CellTypist는 단일세포 전사체 데이터에서 세포 유형을 자동으로 예측하고 주석을 달아주는 Python 기반 패키지입니다. 미리 학습된 reference 모델(예: Immune_All_Low, Immune_All_High, Lung_Immune, Brain_NonImmune, PBMC 등)을 활용하여 각 세포의 발현 패턴을 비교·분류합니다. 사용자는 AnnData 객체를 입력해 간단히 실행할 수 있으며, 결과는 각 세포별 세포 유형 정보로 추가됩니다. 직관적이고 빠른 분류가 가능해 대규모 단일세포 데이터 분석에 유용하며, 연구자의 해석을 보완하는 도구로 널리 활용됩니다.
주요사항
- 모델 목록은 https://www.celltypist.org/models 또는 celltypist.models.models_description() 같은 함수를 통해 확인할 수 있습니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_runCellTypist.py input_dir = ./scanpy_findcluseter/output \ hvg_path = ./scanpy_findhvgs/output \ output_dir = ./scanpy_runcelltypist/output \ model = Immune_All_Low \ majority_voting = TRUE \ over_clustering = FALSE
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_findcluseter/output | 입력 파일이 저장된 디렉토리 경로로, PCA 결과를 포함하는 scanpy_findClusters, scanpy_BBKNNbatchCorrection 등의 결과 폴더임 | |
| Input | Folder | hvg_path | ./scanpy_findhvgs/output | 입력 파일이 저장된 디렉토리 경로로 scanpy_findHVGs 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_runcelltypist/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | model | Immune_All_Low | AnnData 내 세포 유형을 예측하는 데 사용되는 모델. listCellTypistModels 을 통해 얻은 경우 celltypist_models.csv 내 파일 목록에서 .pkl 문자열은 제외하여 작성 | |
| Option | Boolean | majority_voting | True | Over-clustering 후 majority voting을 시행하여, 예측된 라벨링으로 보정할지 여부 | |
| Option | Boolean | over_clustering | False | 이전 clustering 결과에 대해 라벨링할지 여부. False이고 [majority_voting]=True일 경우 입력된 데이터 크기에 따라 heuristic over-clustering 방식을 적용하며, [majority_voting]=False인 경우 이 옵션은 무시됨 |
결과
-
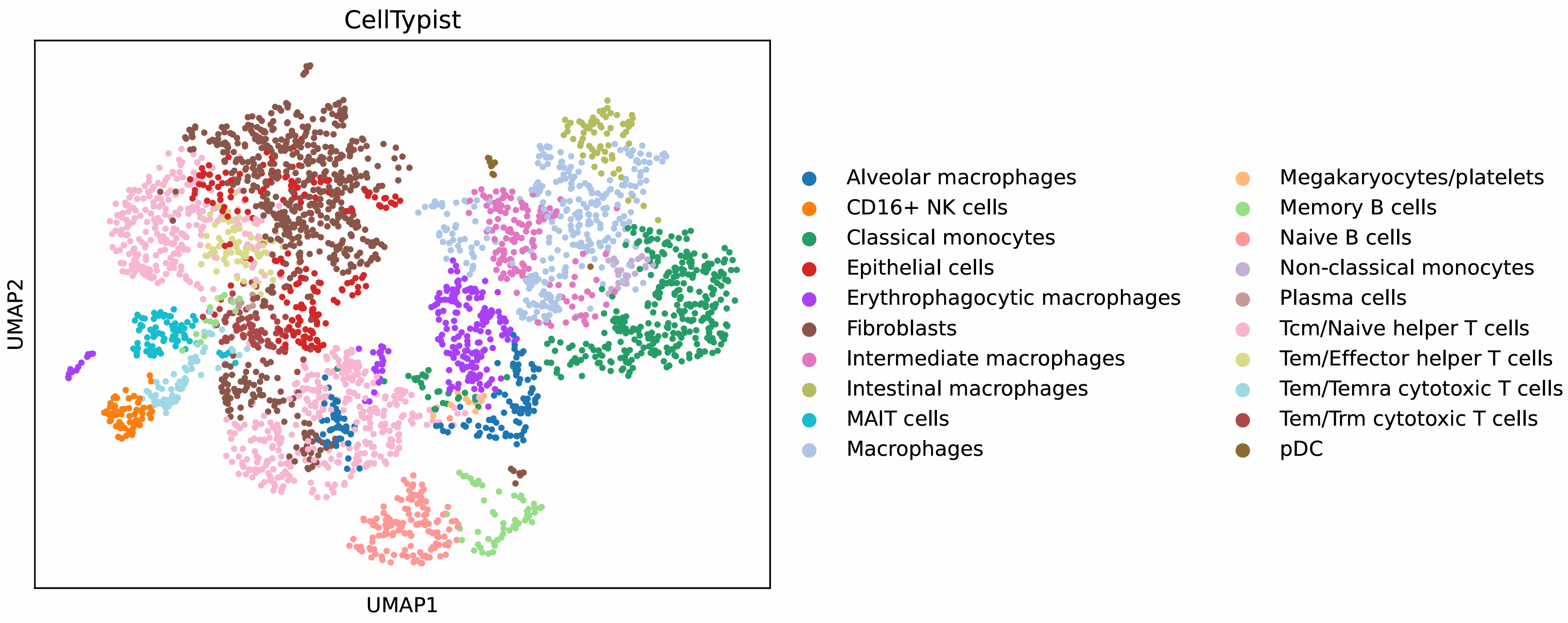
산점도 플롯. scRNA-seq 데이터의 UMAP 결과로, 각 점은 하나의 세포를 나타내고 CellTypist가 예측한 세퍼 유형에 따라 서로 다른 색상으로 구분됨
-

adata_cellType_annotated 파일 내용. 세포 유형 주석이 추가된 AnnData 객체로, 각 세포의 발현 데이터와 클러스터 정보뿐만 아니라, CellTypist를 통해 예측된 세포 유형 정보(obs['CellTypist']가 포함
-

matrix.csv 파일 내용. decision_matrix.csv는 각 세포가 모든 후보 세포 유형에 대해 갖는 의사결정 점수(logit 값) 를 담고 있으며, 값이 클수록 해당 세포 유형일 가능성이 높음을 의미함. probability_matrix.csv는 이 점수를 소프트맥스 변환하여 확률(0~1) 형태로 나타낸 것으로, 각 세포에 대해 모든 세포 유형의 확률 분포를 확인할 수 있음
-

predicted_labels.csv 파일 내용. 각 세포에 대해 최종적으로 할당된 세포 유형(label) 을 기록한 파일임. 여기에는 기본 모델 예측(predicted_labels), over-clustering 기반 그룹 번호(over_clustering), majority voting 보정 결과(majority_voting)가 함께 제공되어, 라벨링 과정을 단계별로 추적할 수 있음
scanpy_findDEG
분석에 사용되는 AnnData 객체를 기반으로, 각 클러스터(그룹) 내 세포와 나머지 세포 간, 혹은 사용자가 지정한 두 클러스터(그룹) 간의 차별 발현 유전자(Differentially Expressed Gene, DEG)를 선별합니다. 이는 두 세포 집단의 유전자 발현량 차이를 Scanpy의 scanpy.tl.rank_genes_groups() 함수에 내장된 t-test, Wilcoxon rank sum test 등의 통계 기법으로 비교하여 수행됩니다. 결과 파일에서는 두 집단 간 발현 차이를 보이는 유전자 목록을 확인할 수 있으며, 이를 시각화하여 활용할 수 있습니다.
주요사항
- 사용자 지정 그룹(groupby)을 기준으로, 각 클러스터 vs 나머지 전체 또는 지정한 두 그룹 간 비교를 수행할 수 있습니다. DEG 선별시에 t-test, wilcoxon (비모수 검정, 기본값으로 널리 사용), logreg(로지스틱 회귀 기반) 등의 다양한 통계방법을 지원하고 있습니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_findDEG.py input_dir = ./scanpy_runcelltypist/output \ output_dir = ./scanpy_finddeg/output \ groupby = leiden \ groups = all \ n_genes = None \ method = t-test \ corr_method = benjamini-hochberg \ tie_correct = FALSE \ pts = FALSE \ pval_cutoff = 0.05 \ log2fc_abs_thr = 0 \ plot_volcano = False \ repel_genes = none \ plot_width = 9 \ plot_height = 6
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_runcelltypist/output | 입력 파일을 담고 있는 디렉토리 경로로 scanpy_findClusters, scanpy_BBKNNbatchCorrection, runcelltyplist, scanpy_runCellTypist, scanpy_annotateClusters 등의 결과 폴더임 | |
| Output | Folder | output_dir | ./scanpy_finddeg/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | groupby | leiden | 차등 발현 분석을 수행하기 위해 세포를 구분하는 그룹(예: 클러스터, 조건) 지정 | |
| Option | String | groups | all | 비교할 대상이 될 [groupby]의 하위 카테고리로 복수 시 공백 없이 쉼표로 구분 (예: 'g1,g2' 또는 'all'(기본값)). [groups]의 첫 번째 항목은 volcano plot을 그릴 때 control로 사용 | |
| Option | Integer | n_genes | none | 반환된 테이블에 나타날 유전자 수로 기본값은 'all' | |
| Option | String | method | t-test | 통계기법 선택. 기본값: 't-test'; 가능한 값: 't-test', 't-test_overestim_var', 'wilcoxon', 'logreg' | |
| Option | String | corr_method | benjamini-hochberg | p-value 보정 방법. 't-test', 't-test_overestim_var', 'wilcoxon'에만 사용. 가능한 값: 'benjamini-hochberg'(기본값), 'bonferroni' | |
| Option | Boolean | tie_correct | False | (Wilcoxon) Wilcoxon 점수에 대해 tie correction을 적용할지 여부 | |
| Option | Boolean | pts | False | 유전자를 발현하는 세포의 비율을 계산할지 여부. | |
| Option | Float | pval_cutoff | 0.05 | 보정된 p-value의 cutoff. 이 값 이하인 유전자만 반환 | |
| Option | Float | log2fc_abs_thr | 0 | 반환할 log2(Fold Change) 절댓값의 threshold. | |
| Option | Boolean | plot_volcano | False | 두 그룹 간의 DEG를 volcano plot을 통해 시각화할지 여부. [groups]의 그룹 수가 2인 경우 또는 [groups]=all이면서 [groupby] 내 카테고리의 수가 2인 경우 True 선택 가능 | |
| Option | Integer | clip_yaxis | 300 | Volcano plot에서 y축 (-log10 p-value) 최댓값. 이 값 이상은 잘림 | |
| Option | String | repel_genes | none | Volcano plot에 표시할 유전자 목록으로 공백없이 쉼표로 구분 | |
| Option | Integer | plot_width | 9 | Volcano plot의 너비 | |
| Option | Integer | plot_height | 6 | Volcano plot의 높이 |
결과
-
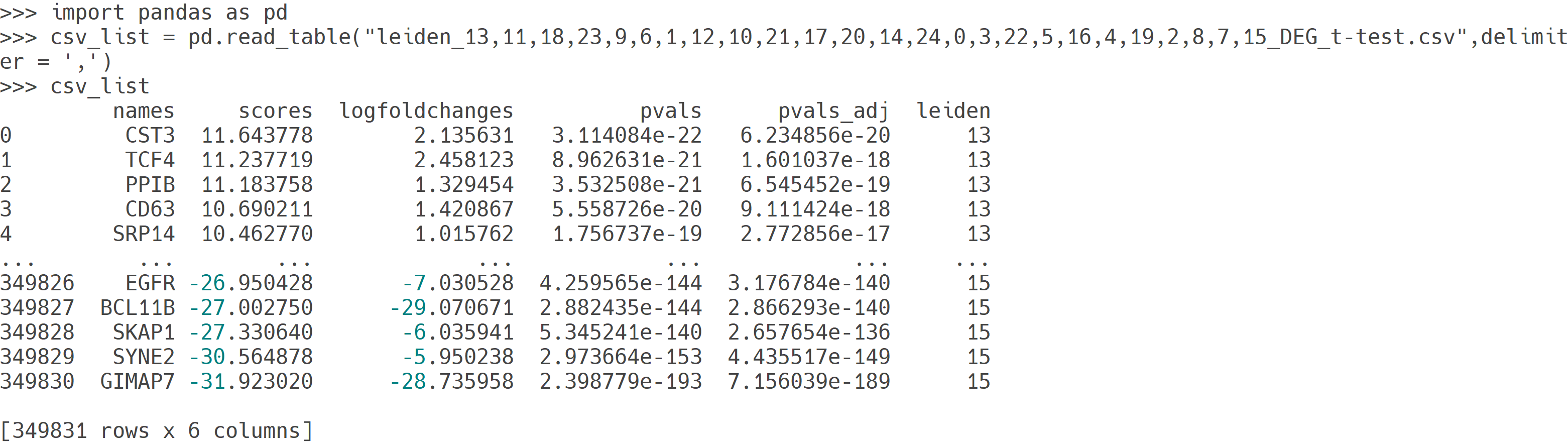
[groupby]_[groups list]_DEG_[method].csv 파일 내용. 이 파일은 클러스터/조건에서 다른 세포 집단과 비교했을 때 발현이 유의하게 달라지는 유전자, 즉 차등 발현 유전자(DEG)를 정리한 것으로 각 유전자에 대해 이름, 통계값, 발현 차이(log2 fold change), 원시 및 보정 p값, 해당 클러스터 ID 등의 정보가 포함
-
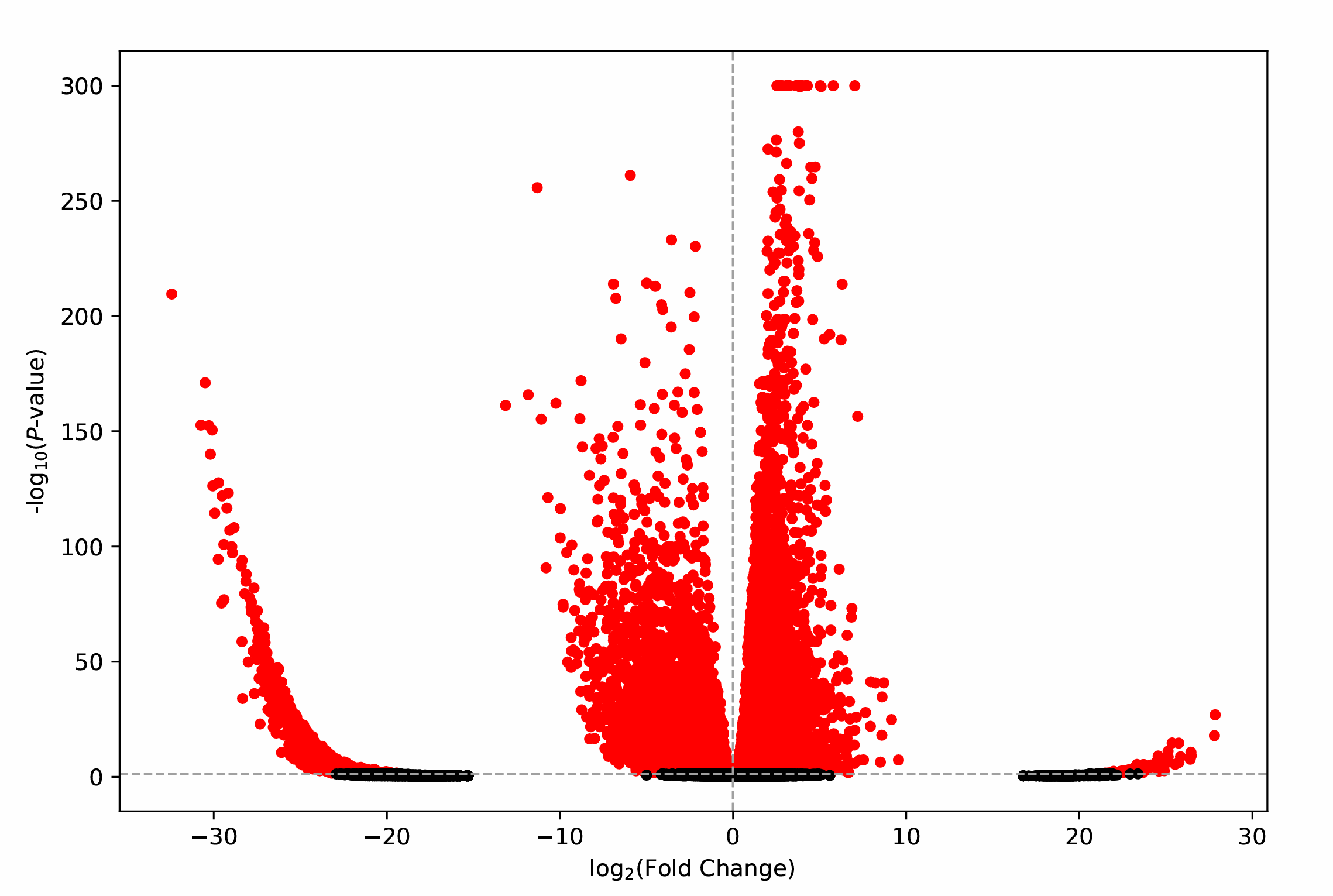
볼케이노 플롯. 두 그룹 간 차등 발현 유전자를 시각화한 그림으로, 각 점은 하나의 유전자를 나타내며 x축은 log2 fold change, y축은 -log10 보정 p값을 의미함. 이 그림은 그룹 수가 2개일 때만 생성 가능하므로, 여기서는 sample_id를 기준으로 두 그룹 간 발현 차이를 비교하여 DEGs를 시각화함
scanpy_plotGeneExpr
scanpy_plotGeneExprs 함수는 사용자가 지정한 유전자 발현을 scatter plot, dot plot, violin plot, 혹은 heatmap 형태로 시각화하는 기능을 제공합니다. 이 함수는 DEG(차등 발현 유전자)를 계산하지 않으며, 사용자가 직접 지정한 유전자 목록이나 사전에 수행된 DEG 분석 결과를 기반으로 발현 양상을 시각화합니다. 출력된 그래프는 자동으로 파일로 저장되며, 연구자는 이를 통해 각 세포 집단 간 발현 패턴을 직관적으로 비교하고 해석할 수 있습니다. 이러한 시각화는 후속 분석이나 결과 보고에 매우 유용하게 활용됩니다.
주요사항
- input_dir 내부에 다음 파일 중 하나가 반드시 존재해야 합니다. 우선 annotation 결과인 adata_cellType_annotated.h5ad이 로딩되며, 파일이 없을 경우는 adata_scaled.h5ad파일이 로딩됩니다. groupby에 입력하는 값은 anndata의 obs 내에 column이 있어야 하며, features 유전자 목록을 입력시에 "Cd3e,Pdcd1,Lag3" 처럼 콤마로 구분된 문자열을 입력해야 하며, 공백 없이 써야 합니다. "none"을 입력하면 자동으로 DEG를 계산해서 상하위 3개씩 유전자를 선택합니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_plotGeneExprs.py input_dir = ./scanpy_runcelltypist/output \ output_dir = ./scanpy_plotGeneExprs/output \ feature = none \ plot_type = scatter \ scatter_basis = umap \ dendrogram = False \ stack_violin =False \ groupby = leiden \ layer = normalized/ scaled \ thres = none \ save = .pdf
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_runcelltypist/output | 입력 파일을 담고 있는 디렉토리 경로 | |
| Output | Folder | output_dir | ./scanpy_plotGeneExprs/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | feature | none | 쉼표로 구분된 문자열로 그릴 유전자 목록을 지정. (예: Pdcd1,Lag3) None이면 각 카테고리당 3개의 up/downregulated gene이 표시됨 | |
| Option | String | plot_type | scatter | 유전자 발현을 시각화할 plot 유형 지정. [가능한 값: “scatter”, "dot", "violin", "heatmap"] | |
| Option | String | scatter_basis | umap | ([plot_type]이 scatter인 경우) 시각화에 사용할 임베딩 선택. 기본값: "umap"; 가능한 값: "umap", "tsne", "pca" | |
| Option | Boolean | dendrogram | False | ([plot_type]이 dot/violin/heatmap인 경우) True인 경우, [groupby] 내 hierarchical clustering에 기반한 dendrogram 추가 | |
| Option | Boolean | stack_violin | False | ([plot_type]이 violin이고 [feature] 내 유전자가 2개 이상인 경우) - True: 여러 개의 violin plot을 하나의 plot 안에 시각화 - False: 유전자별로 시각화 | |
| Option | String | groupby | leiden | ([plot_type]이 dot/violin/heatmap인 경우) 시각화 시 세포를 구분하는 그룹(예: 클러스터, 조건)을 지정 | |
| Option | String | layer | normalized/ scaled | 시각화할 AnnData 내 expression matrix의 type. [가능한 값: "raw", "normalized", "scaled"] - [plot_type]이 scatter, violin: 기본값으로 log-normalized expression이 시각화됨. - [plot_type]이 dot, heatmap: 기본값으로 z-scaled expression이 시각화됨 | |
| Option | String | thres | none | ([layer]이 scaled 이고 [plot_type]이 dot 또는 heatmap인 경우)) 시각화할 발현값의 절댓값 cutoff 지정 | |
| Option | String | save | 출력할 그림 파일의 확장자 지정. 기본값: ".pdf" |
결과
-
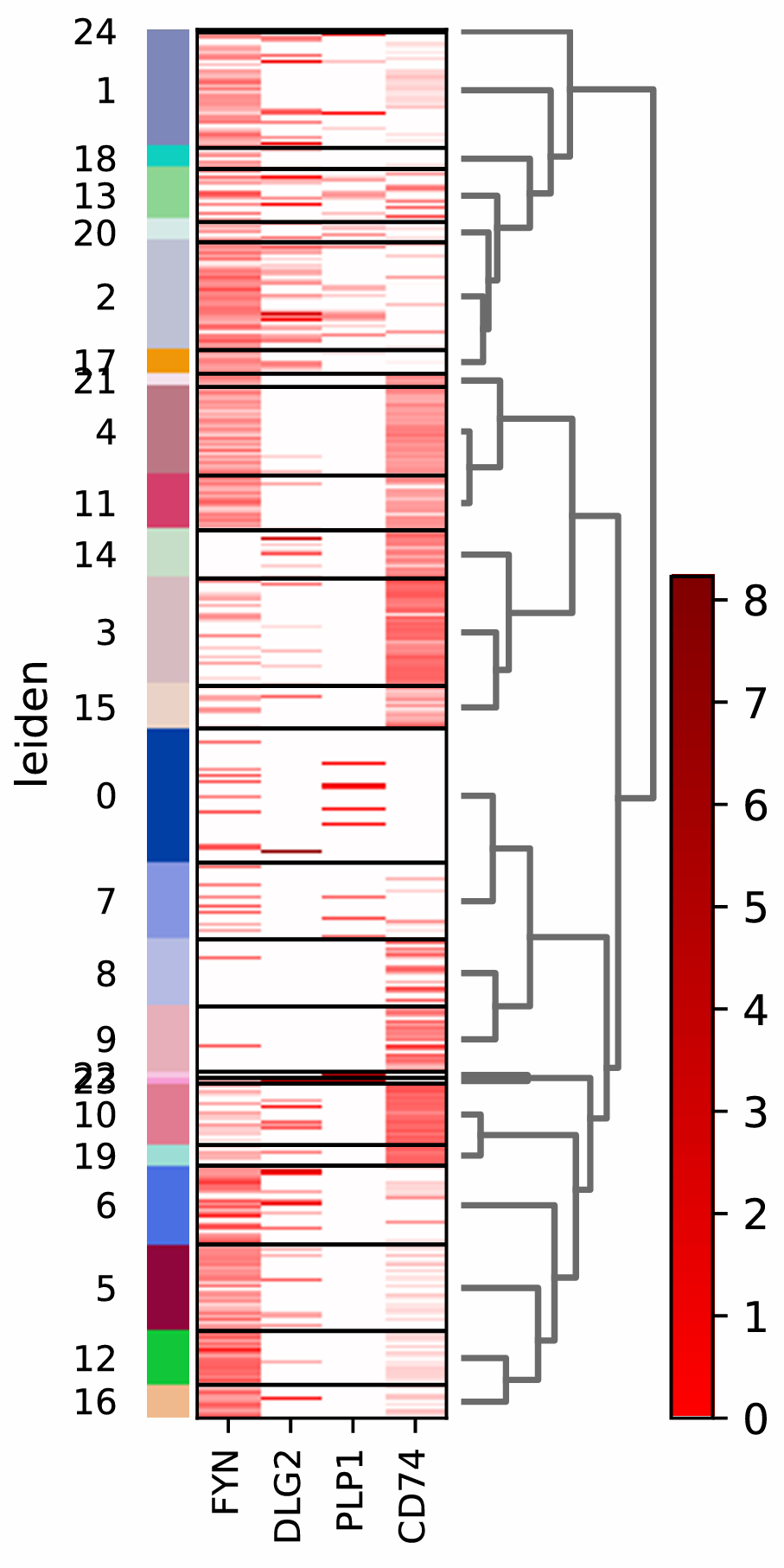
히트맵. 특정 유전자 (예: LYZ, CST3, MALAT1, CD3E)의 z-scaled 발현을 색상으로 한 눈에 비교
-
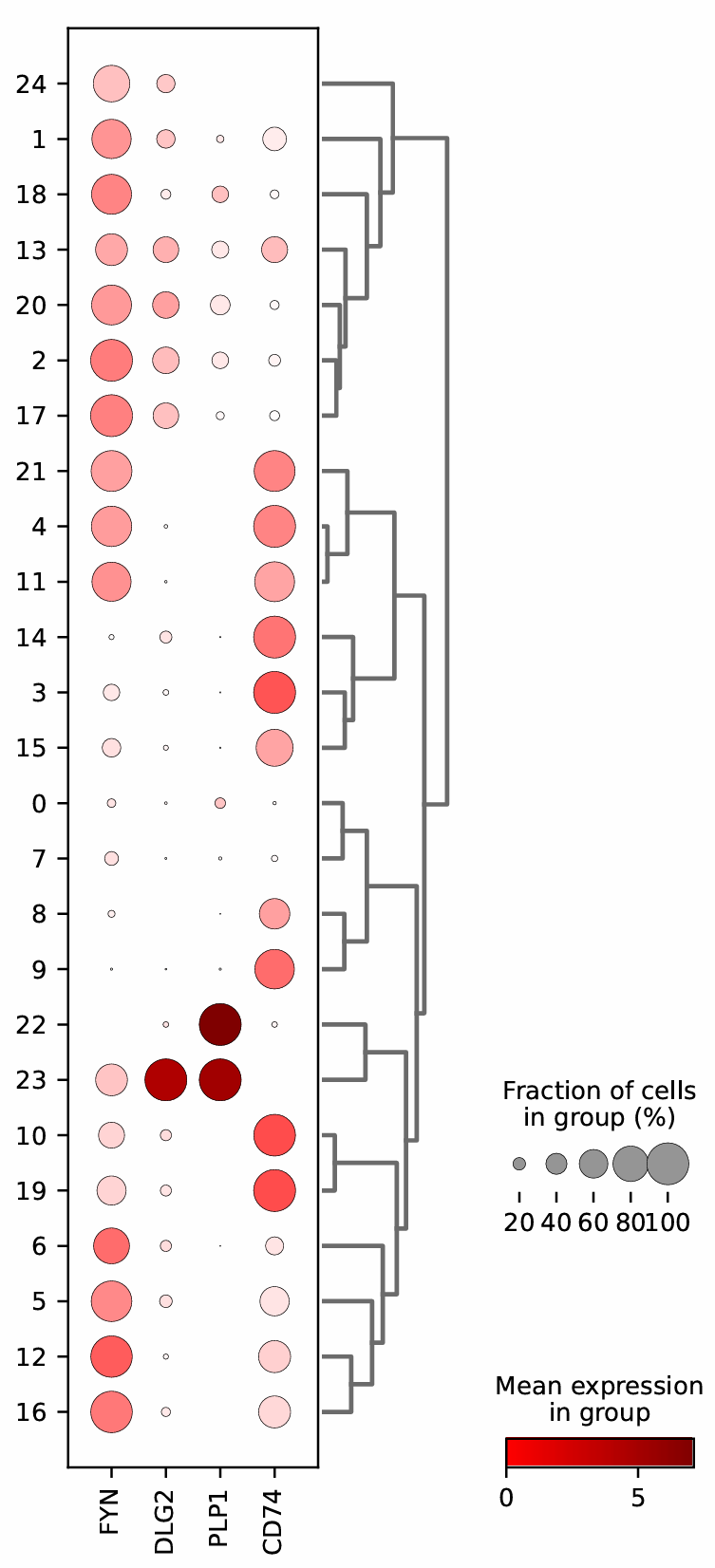
닷 플롯. 특정 유전자 (예: LYZ, CST3, MALAT1, CD3E)의 z-scaled 발현 수준을 점 크기와 색상으로 시각화
-
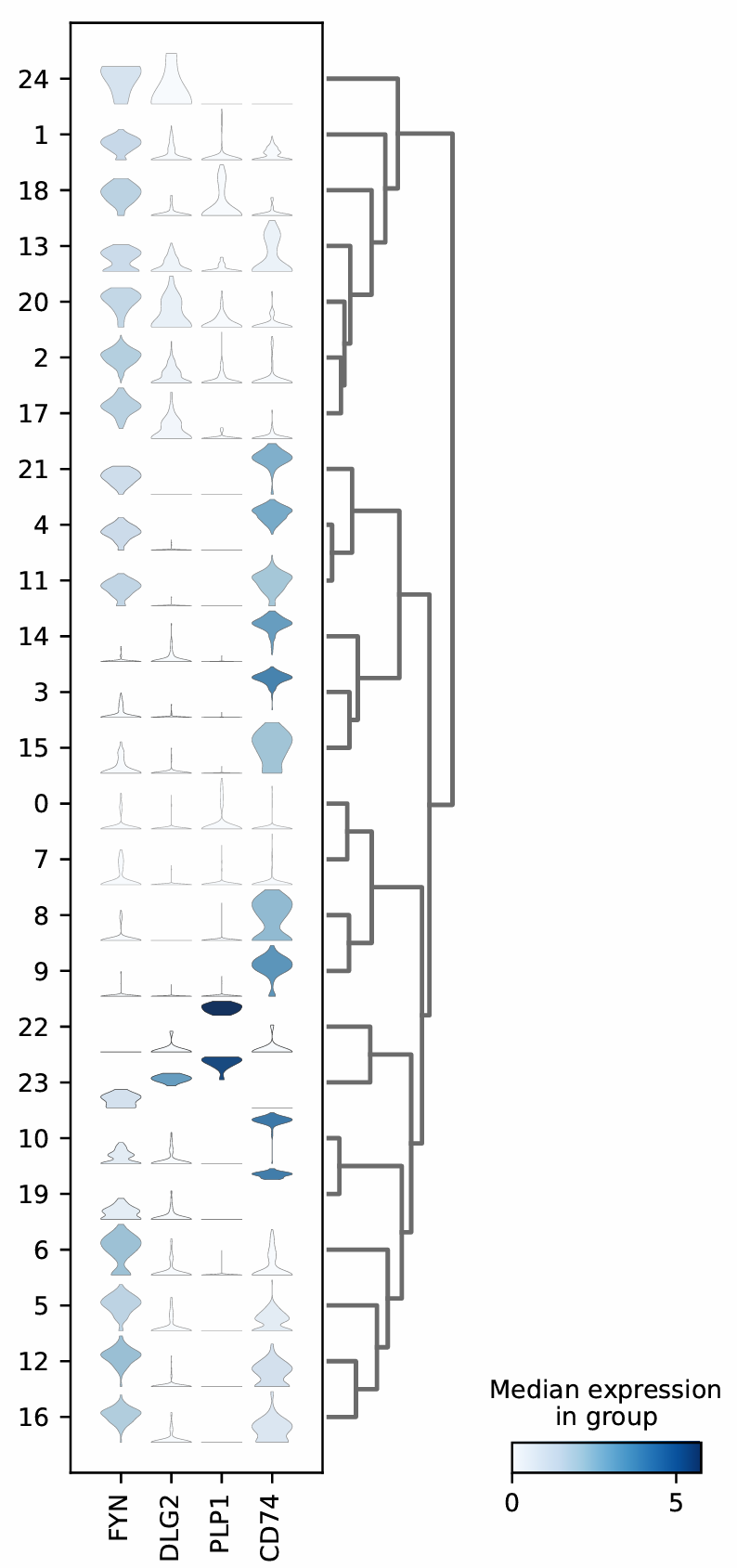
누적 바이올린 플롯. 특정 유전자 (예: LYZ, CST3, MALAT1, CD3E)의 log-normalized 발현 분포를 층별로 비교
-
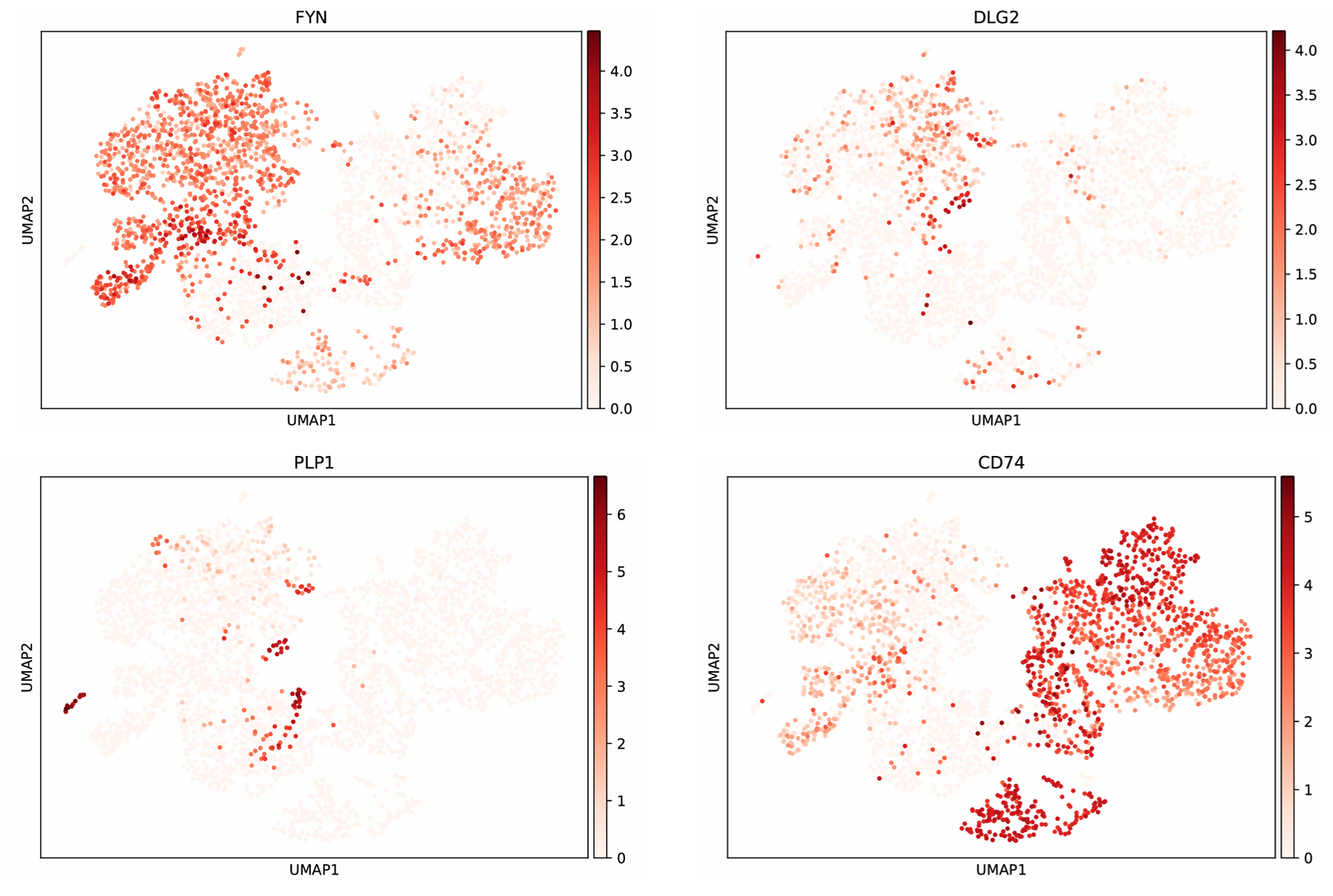
산점도 플롯. 특정 유전자 (예: LYZ, CST3, MALAT1, CD3E)의 log-normalized 발현 분포와 수준(level)을 세포 단위로 확인
scanpy_plotCellTypeProportion
scanpy_plotCellTypeProportion 함수는 AnnData 객체 내 두 개의 범주형 변수(예: 클러스터와 세포 유형)를 기반으로 교차 빈도표(contingency table)를 생성합니다. 이를 통해 첫 번째 그룹의 각 카테고리(예: 세포유형)에 속한 세포들이 두 번째 그룹(예: 클러스터)의 각 카테고리에 얼마나 포함되는지 계산할 수 있습니다. 계산된 값은 절대 빈도(absolute counts)와 상대 점유율(relative proportions)로 제공되며, 이를 막대그래프로 시각화하여 각 클러스터에서 어떤 세포 유형이 어느 정도 분포하는지 직관적으로 확인할 수 있습니다.
주요사항
- AnnData.obs에 존재하는 두 범주형(categorical) 변수(group1 = 클러스터, group2 = celltype)를 기준으로 교차 빈도표(contingency table)를 생성합니다. 클러스터 내에서 celltype의 카테고리(예: 세포 타입)가 차지하는 비율을 계산하고, 절대값(빈도)과 상대값(점유율)을 모두 제공합니다. 이를 통해 각 클러스터(leiden)에서 어떤 세포 타입(celltype)이 얼마나 분포되어 있는지 시각화할 수 있습니다.
입력 데이터 예제
실행 명령어 예시
python scanpy_plotCellTypeProportion.py input_dir = ./scanpy_runcelltypist/output \ output_dir = ./scanpy_plotcelltypeportion/output \ group1 = sample_id \ group2 = CellTypist \ plot_width = 9 \ plot_height = 6
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_runcelltypist/output | 입력 파일을 담고 있는 디렉토리 경로 | |
| Output | Folder | output_dir | ./scanpy_plotcelltypeportion/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | group1 | sample_id | AnnData 내 첫 번째로 세포를 구분하는 그룹(예: 클러스터, 조건)을 지정 | |
| Option | String | group2 | CellTypist | AnnData 내 두 번째로 세포를 구분하는 그룹(예: 클러스터, 조건)을 지정 | |
| Option | Integer | plot_width | 9 | Bar plot의 너비 | |
| Option | Integer | plot_height | 6 | Bar plot의 높이 |
결과
-
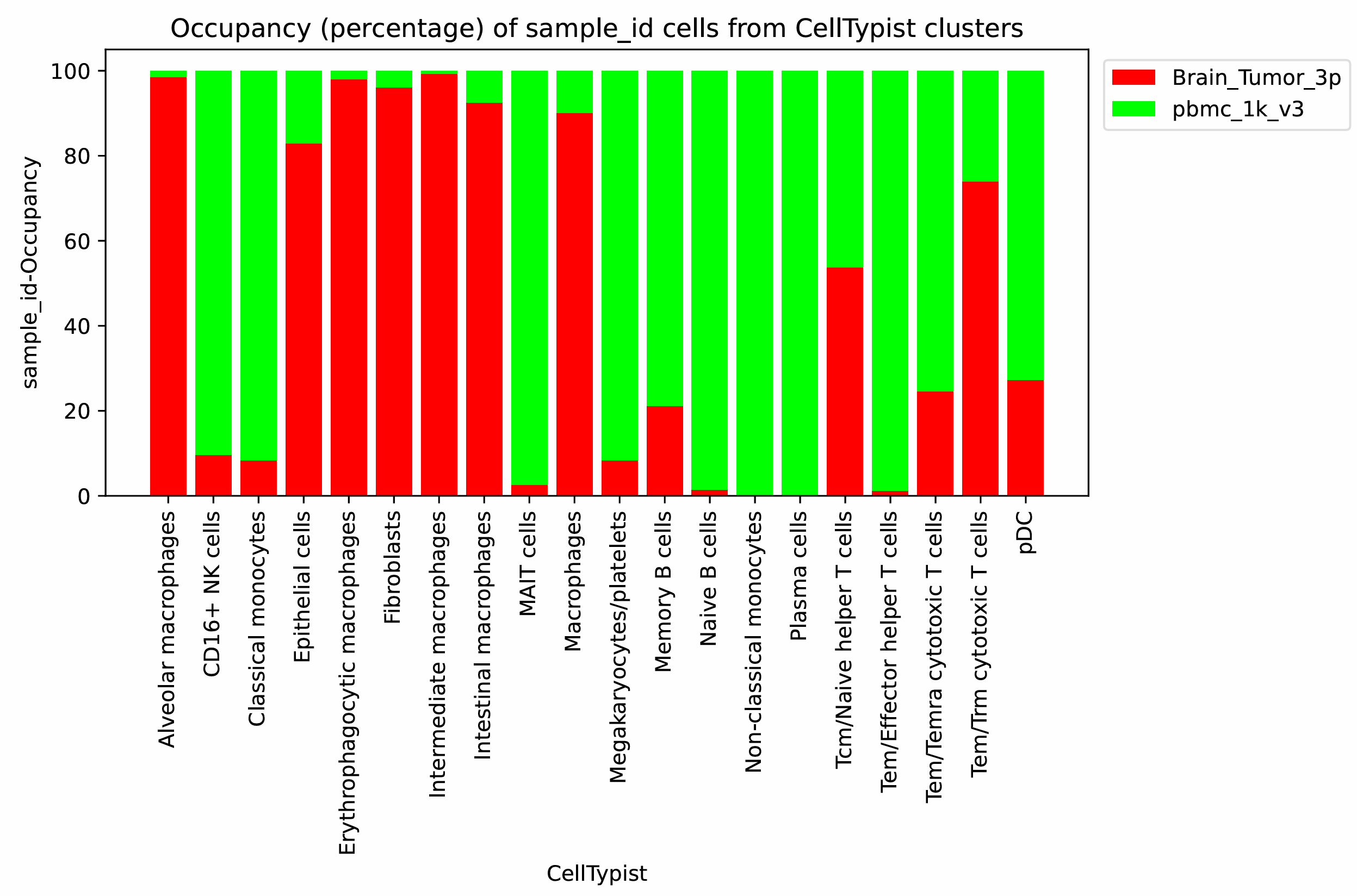
상대 점유율 표에 대한 막대 플롯. Group2 (예: sample_id) 에서 예측된 Group1 (예: CellTypist)별 세포 비율을 시각화
-
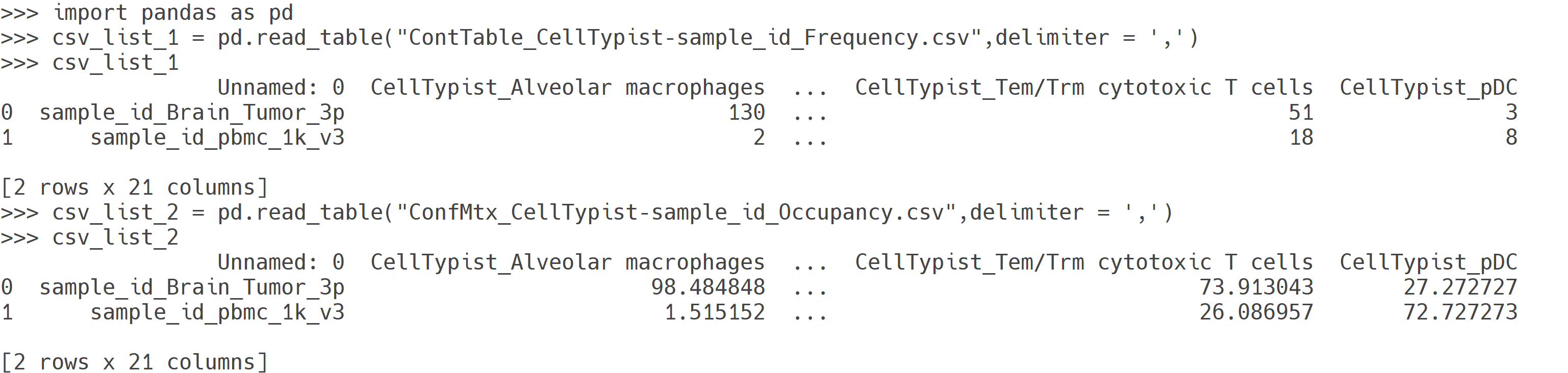
[ContTable/ConfMtx]_[group1]_[group2]_[Frequency/Occupany].csv 파일 내용. Group2 (예: sample_id) 에서 예측된 Group1 (예: CellTypist)별 세포 분포 및 비율을 정리한 표
-
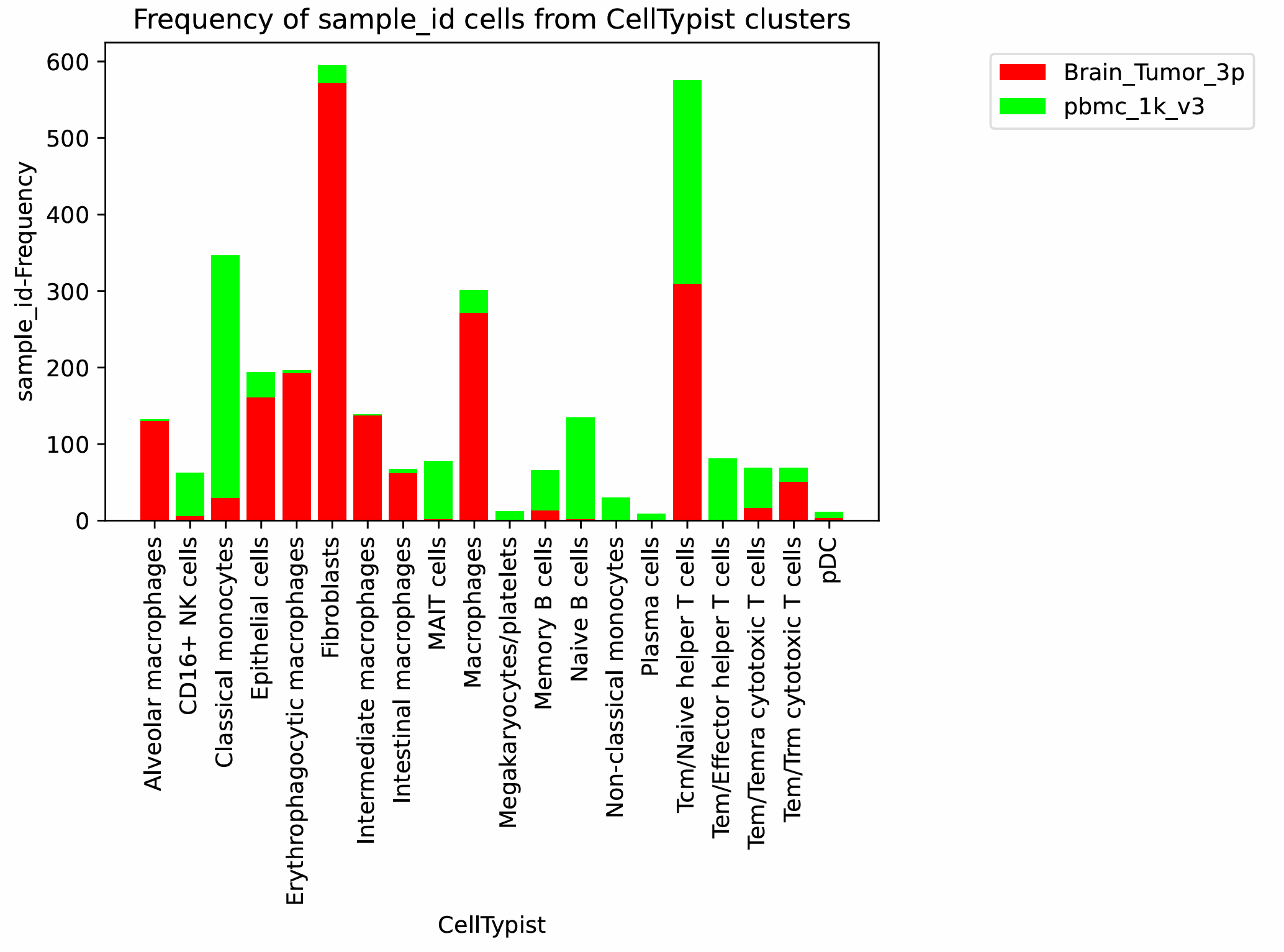
절대 빈도 표에 대한 막대 플롯. Group2 (예: sample_id) 에서 예측된 Group1 (예: CellTypist)별 세포 분포를 시각화
scanpy_extractGenes
scanpy_extractGenes.py는 Scanpy를 이용한 전처리 및 정규화 과정 후, 분석에 사용되는 전체 유전자 목록을 추출하는 프로그램입니다. 이 스크립트는 특히 topGO 분석과 같은 하위 집합 기반 유전자 기능 분석에 필요한 입력 파일을 준비하는 과정에서 사용됩니다. 사용자는 먼저 scanpy_normalize를 통해 정규화된 AnnData 객체를 준비하고, 이를 기반으로 본 프로그램을 실행하여 전체 유전자의 리스트를 추출합니다. 추출된 유전자 리스트는 각 유전자에 대한 식별자 정보와 함께 CSV 또는 텍스트 파일 형태로 저장되며, 이후 topGO의 입력값으로 활용할 수 있습니다.
주요사항
- 관심 유전자 리스트를 기준으로 adata[:, genes]로 서브셋팅하며, 중복·대소문자·ID 매핑을 정리해 일관성을 확보한다. 정규화·로그 처리된 데이터는 adata.raw에서 추출해야 재현성이 보장되고, sc.tl.rank_genes_groups로 차등발현을 확인한 뒤 to_df().to_csv()로 결과를 저장한다.
입력 데이터 예제
실행 명령어 예시
scanpy_extractGenes.py input_dir = ./scanpy_nomalize/output \ output_dir = ./scanpy_extractgenes/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_nomalize/output | 입력 파일을 담고 있는 디렉토리 경로 | |
| Output | Folder | output_dir | ./scanpy_extractgenes/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | test | test | 테스트 |
결과
-
.png)
detected_gene_list.csv 파일 내용. scanpy_normalize 결과로부터 추출된 전체 유전자 목록
topGO_GOanalysis
TopGO는 특정 유전자 목록을 기반으로 Gene Ontology(GO) 용어의 풍부도를 분석하는 R 패키지입니다. 이를 통해 차이가 있거나 관심 있는 유전자들이 GO의 어떤 기능 범주(Biological Process, Molecular Function, Cellular Component)에 과도하게 포함되어 있는지를 통계적으로 평가할 수 있습니다. GO의 계층 구조를 고려하여 부모-자식 노드 간 의존성을 반영한 보다 정확한 enrichment 분석이 가능하며, Fisher’s exact test, Kolmogorov-Smirnov test, weighted scoring 등 다양한 통계적 방법을 지원합니다.
주요사항
- topGO는 차등 발현 유전자(DEGs) 리스트를 이용하여 GO(Gene Ontology) 기반의 기능적 풍부도 분석(functional enrichment analysis) 을 수행하는 R 기반 패키지입니다. 이 도구는 각 유전자의 GO term 간의 구조적 관계(GO hierarchy)를 고려하여, 단순한 과대표현(over-representation) 분석보다 더 정교한 통계적 유의성 평가를 수행합니다. 분석 시 Fisher’s exact test, Kolmogorov–Smirnov test 등 다양한 통계 검정을 지원하며, ‘elim’, ‘weight’, ‘classic’ 등의 알고리즘을 통해 GO term 간 종속성을 보정합니다. 기본적으로 분석 대상 생물종은 인간(hsapiens) 으로 설정하였고, 분석 소스는 GO:BP (Biological Process) 로 설정하였습니다.
입력 데이터 예제
실행 명령어 예시
Rscript topGO_GOanalysis.r \ input_dir = ./scanpy_finddeg/output \ gene_list_dir = ./scanpy_extractGenes/output \ output_dir = ./topgo_goanalysis/output \ species = human \ deg_pval_cutoff = 0.05 \ deg_logfc_abs_thr = 0.5 \ subont = BP \ p_cutoff = 0.05 \ plot = True \ groupby = leiden \ plot_go_id = FALSE \ plot_width = 16 \ plot_height = 9
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_finddeg/output | 차등 발현 유전자(DEG) 목록 파일들이 저장되어 있는 디렉토리 경로 | |
| Input | Folder | gene_list_dir | ./scanpy_extractGenes/output | 분석에서 참조로 사용할 전체 유전자 목록 파일들이 저장되어 있는 디렉토리 경로 | |
| Output | Folder | output_dir | ./topgo_goanalysis/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | species | human | 현재 "mouse"(Mus musculus)와 "human"(Homo sapiens)을 지원함 | |
| Option | Float | deg_pval_cutoff | 0.05 | GO 분석에 사용할 계산된 DEG의 조정된 P-value의 임계값 지정 | |
| Option | Float | deg_logfc_abs_thr | 0.5 | GO 분석에 사용할 계산된 DEG의 조정된 logFC 절댓값의 임계값 지정 | |
| Option | String | subont | BP | Gene Ontology의 서브 온톨로지. "BP"(Biological Process), "CC"(Cellular Component), "MF"(Molecular Function) 중 선택 | |
| Option | Float | p_cutoff | 0.05 | 계산된 Gene Ontology의 P-value(Fisher's exact test 사용) 임계값으로 이 값 이하의 항목은 저장되지 않음 | |
| Option | Boolean | plot | TRUE | 계산된 GO term에 대한 막대그래프를 저장할지 여부. 상위 5개의 up/downregulated term 포함 | |
| Option | String | groupby | leiden | 세포를 구분하는 그룹(예: 클러스터, 조건) 지정 | |
| Option | String | plot_go_id | FALSE | 막대그래프 축 눈금에 GO ID 사용 여부 | |
| Option | Integer | plot_width | 16 | 그래프 파일의 가로 길이 | |
| Option | Integer | plot_height | 9 | 그래프 파일의 세로 길이 |
결과
-
.png)
GO[subont]_[Down/Up]_reg_[groupby]-[group name].csv 파일 내용. 특정 클러스터([groupby]-[group name])에서 상향 또는 하향 조절된 유전자와 관련된 GO 항목의 ID, 용어, 주석된 유전자 수, 유의한 유전자 수, 기대값, Fisher.elim p-value를 포함한 표 형식 데이터
-
.png)
BarPlot_GO[subont]_[groupby]-[group name].png 파일. topGO 분석 결과(csv)에서 클러스터별 상향(Up) 및 하향(Down) 조절 유전자와 관련된 GO 항목 중 상위 5개를 추출하고, -log10(p-value) 기준으로 정렬하여 막대그래프 형태로 시각화한 PNG 파일
scanpy_subsetAnnData
scanpy_subsetAnnData는 특정 그룹 내 특정 카테고리의 세포들에 대해 raw UMI count matrix를 분리하고, 새로운 AnnData로 저장합니다. Subset 후 Normalization과 Scaling을 다시 수행하려면 원본 데이터(raw matrix)를 사용해야 합니다. 이미 가공된 데이터를 기반으로 subset을 수행하면, 기존 통계 정보가 남아 있어 새로운 subset에 적합하지 않은 결과가 나올 수 있습니다. 따라서 adata_qc_passed_concat.h5ad에 있는 가공되지 않은 원본 데이터로 subset을 수행해야 정확한 분석이 가능합니다. 이 프로그램은 특정 세포 유형(예: T 세포)이나 조건별 하위 집단만 별도로 분석하고자 할 때 사용되며, 원하는 subset을 추출한 뒤 해당 데이터에 맞추어 정규화, 스케일링을 포함한 후속 분석 전 과정을 다시 수행할 수 있습니다.
주요사항
- adata_cellType_annotated.h5ad 파일만으로 subset이 가능합니다. subset을 진행한 이후에도 normalization 또는 scaling 등 전처리를 다시 수행할 필요가 없으며 이미 .layers["raw"], .X, .obs, .var가 적절히 포함되어 있습니다. 다만, subset 후 정규화를 다시 하거나, raw matrix 기반 분석(DEG 등)을 처음부터 할 예정인 경우는 기존 .X가 scaled matrix로 되어 있고 .raw 또는 .layers["raw"]가 비어 있기 때문에 adata_qc_passed_merged.h5ad가 필요합니다. 그렇기 때문에 adata_cellType_annotated.h5ad만 사용해 subset을 수행하되, 사용자 필요 시 adata_qc_passed_concat.h5ad에서 raw matrix를 복구하여 .layers["raw"] 또는 .raw에 설정하는 구조로 변경하였습니다.
실행 명령어 예시
python scanpy_subsetAnnData.py \ input_dir = ./scanpy_runcelltypist/output \ merged_in = ./scanpy_applyqcthresholds/output \ output_dir = ./scanpy_subsetanndata/output \ groupby = CellTypist \ groups = "Classical monocytes"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./scanpy_runcelltypist/output | adata_cellType_annotated.h5ad 등의 입력 파일이 저장된 디렉토리 경로 | |
| Input | Folder | merged_input_dir | ./scanpy_applyqcthresholds/output | QC 후 가공되지 않은 AnnData object(adata_qc_passed_concat.h5ad)가 저장된 디렉토리 경로 | |
| Output | Folder | output_dir | ./scanpy_subsetanndata/output | 결과 파일이 저장될 디렉토리 경로 | |
| Option | String | groupby | CellTypist | 추출하고자 하는 세포의 AnnData object 내 변수(컬럼) 이름 | |
| Option | String | groups | "Classical monocytes" | 추출하고자 하는 세포의 AnnData object 내 변수(컬럼)의 특정 값. 다수일 경우 쉼표로 구분 |
결과
-
.png)
adata_subset_CellTypist_Classical monocytes.h5ad 파일 내용. 특정 그룹/카테고리의 세포를 추출하여 생성한 AnnData 객체
- 버전2.0
- 마지막 업데이트43일 전
- 기여자
- 김
- 이
- 더
- 클
- 차
- 유
- 이
- 장
- 최
- 백
- cellranger_mkref
- cellranger_count
- dropletUtils_filterCells
- scanpy_applyQCthresholds
- scanpy_normalize
- scanpy_findHVGs
- scanpy_scaleData
- scanpy_runPCA
- scanpy_findNeighbors
- scanpy_runTSNE
- scanpy_runUMAP
- scanpy_findClusters
- scanpy_scatterPlot
- scanpy_BBKNNbatchCorrection
- scanpy_annotateClusters
- scanpy_runCellTypist
- scanpy_findDEG
- scanpy_plotGeneExpr
- scanpy_plotCellTypeProportion
- scanpy_extractGenes
- topGO_GOanalysis
- scanpy_subsetAnnData