Korea Bioinformation Center
National center for biological research resources and informationKOBIC News

- 2025-09-01
- 2025-07-24
Public Analysis Pipelines
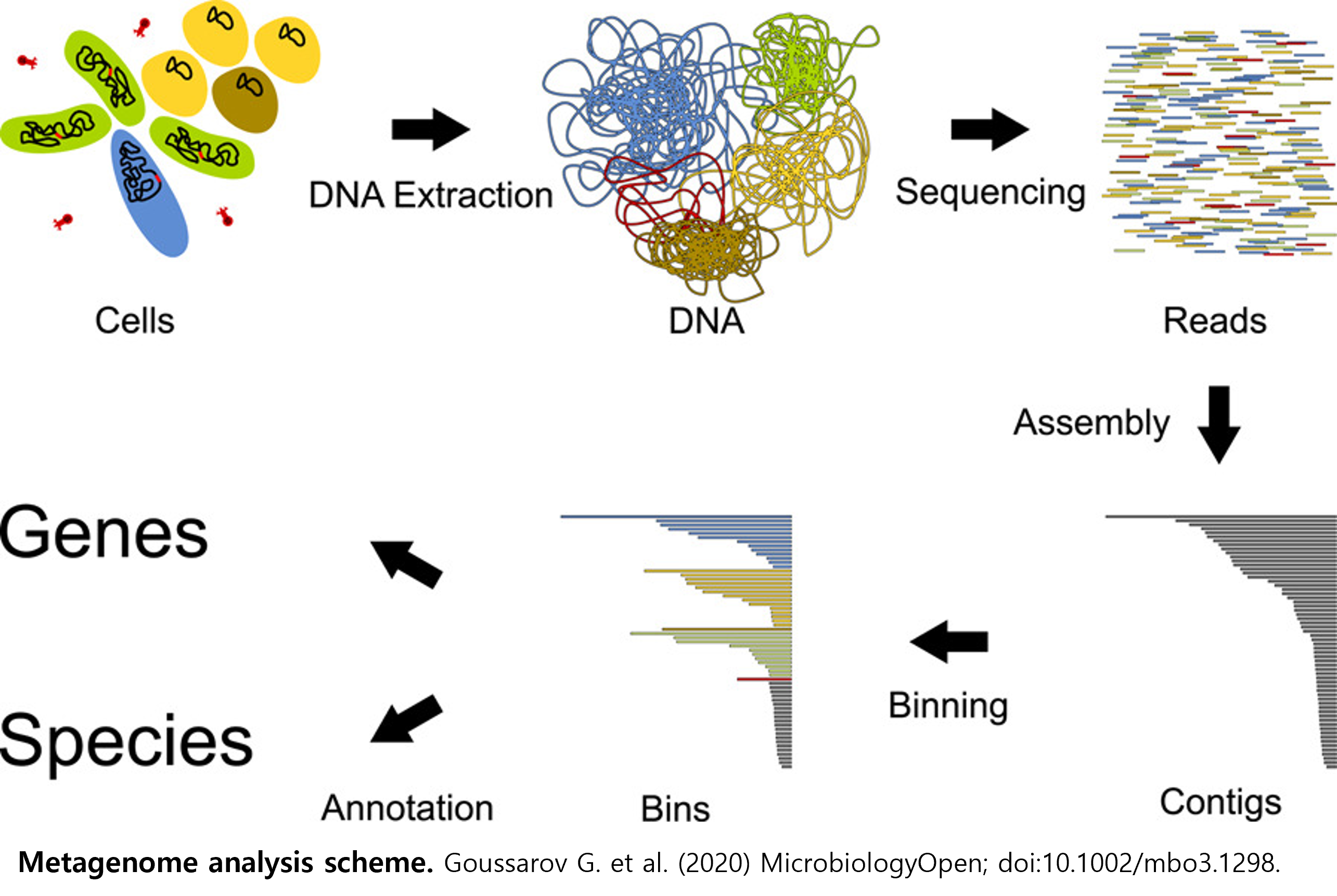

.png)
The BioExpress service is the only cloud-based integrated data analysis service in Korea that enables big data analysis in scientific fields through a dynamic container-based automated workflow analysis platform and high-speed data transfer service.
Download
Please download the workbench and high-speed transfer service for the OS that matches your environment.- CLOSHA : Cloud-based Large-scale Genomic Analysis Platform - GBOX : High-speed transfer service for large amounts of data - SFTP : SSH Protocol-based Data Transfer Service
6,681
User94,791cases
Execution TaskBio research data refers to all types of data produced through national R&D projects in the life sciences field. As innovative research methods utilizing this data gain attention, bio data is emerging as a key factor driving R&D innovation. To support this, the National Bio Data Station has been established to integrate and provide data scattered across ministries, projects, and researchers, aiming to create a data-driven bio research environment.
Registration Status by Data Type
-
2,433cases
Bio Project -
163,038cases
Bio Sample -
2,393,058cases
Registered Data
Bio Project Registration Status
Cumulative Number of Registrations(cases)
Bio big data, which forms the foundation of precision medicine, is becoming increasingly important as the focus shifts from post-treatment care to personalized treatment and preventive healthcare. Particularly in the bioindustry, which benefits from first-mover advantages, proactive investment is necessary, and major countries are building large-scale bio big data systems. Therefore, this project was launched to build national bio big data for leading future healthcare. The goal is to establish a foundation at the national level to collect, store, and utilize 'bio big data', the center of the precision medicine era, and to contribute to the promotion of new industries and the improvement of healthy lives.
Designating and operating 16 rare disease collaboration institutions to recruit rare disease patients and collect clinical information.
Transporting the collected rare disease patients' samples to resource production institutions for the production and analysis of genomic data.
The collected clinical information and genomic data are shared through a consortium formed by three institutions.
The analyzed data is used for rare disease patient counseling, diagnosis, and research activities.
Data Status
The Infectious Disease Data Portal is a portal service that integrates and provides research data on infectious disease viruses from around the world.In a rapidly changing environment, to understand infectious diseases and develop treatments and vaccines, KOBIC integrates and provides global infectious disease research data to share data and results harmoniously.
Sequence Dashboard
Provides integrated information on viruses including disease overview, particle and genomic structure, life cycle, epidemiology, mutations, etc.
Provides quality-analyzed genomic and protein sequences, and protein structures collected from around the world.
Offers various statistical services on virus data, such as outbreak timing, region, mutations, etc.
Simple web-based BLAST service for infectious disease standard genomic sequences.